Redacción Farmacosalud.com
Para la Dra. Francina Munell, neuropediatra del Hospital Universitario Vall d'Hebron (Barcelona), cuando se habla de Atrofia Muscular Espinal (AME) resulta enormemente “importante hacer el diagnóstico precoz y empezar el tratamiento” lo más rápidamente posible, puesto que “una demora de 2-3 semanas puede causar un cambio significativo en la respuesta” a las nuevas terapias que han aparecido para esta enfermedad. Munell realizó estas declaraciones en el marco del webinar formativo ‘6 preguntas prácticas para el diagnóstico y abordaje de la AME’, en el que también participaron como ponentes la Dra. y neuropediatra Inmaculada Pitarch, del Hospital Universitario Politécnico la Fe (Valencia), y la Dra. Julita Medina, médica rehabilitadora del Hospital Universitario Sant Joan de Déu Barcelona (Esplugues de Llobregat, en Barcelona), mientras que como moderador estuvo presente el Dr. Marcos Madruga, neuropediatra del Hospital Viamed Santa Ángela de la Cruz (Sevilla). Cada ponente contestó a dos preguntas del total de seis interrogantes planteados. La sesión, patrocinada por Biogen, Novartis y Roche, contó con el aval científico de la Sociedad Española de Neurología Pediátrica (SENEP).

Fuente: www.farmacosalud.com
Dra. Francina Munell
1- ¿Cuáles son las bases genéticas de la AME?
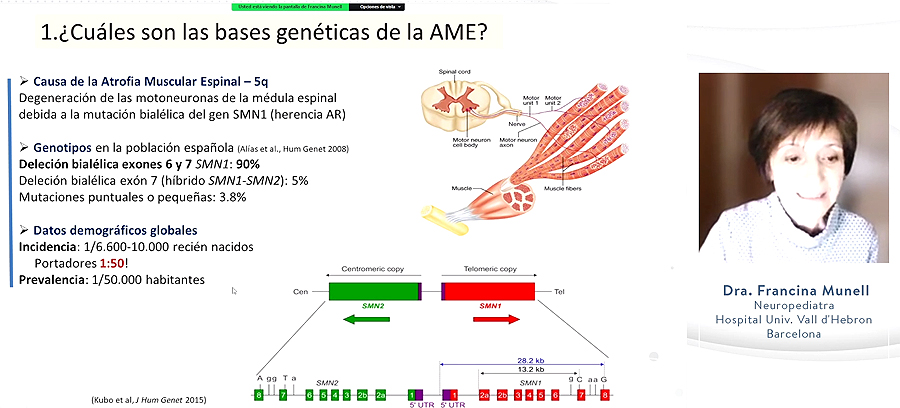
La Atrofia Muscular Espinal (AME) es una enfermedad rara, hereditaria y monogénica, es decir, hay una mutación en un gen determinado, en este caso el SMN1, que produce una degeneración de las motoneuronas de la médula espinal. Dado que es una patología ligada a la herencia autosómica recesiva, se necesitan tener mutadas las dos copias del gen, los dos alelos, los recibidos por vía materna y paterna. Si sólo se presenta alteración en una de las copias, la persona es portadora asintomática.
En el ámbito de la AME, lo más típico es que la mutación se configure como una deleción de las dos copias, pudiendo ser de un exón o de dos exones. La alteración más frecuente es la deleción de los exones 6 y 7 en el SMN1, que representa el 90% de las causas de AME en la población española. En un 5% puede haber deleción de un solo exón, y en menos de un 5% se registran mutaciones puntuales o pequeñas.
Difusión: www.farmacosalud.com
Un aspecto importante de la AME es el que gira en torno a la intervención de un segundo gen, el denominado SMN2, ya que “nos puede modificar la severidad de la enfermedad”, afirmó la Dra. Munell. El SMN2, que se caracteriza por producir escasa proteína, puede presentar varias copias. Evidentemente, cuantas más copias haya, más proteína se obtendrá y, por lo tanto, cuantas más copias de este gen estén presentes, más leve será la patología. Las copias de SMN2 “son el principal modificador” de la AME, subrayó Munell.
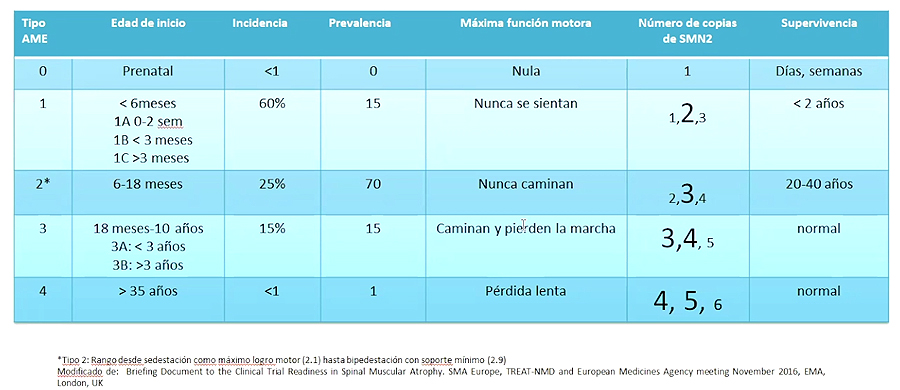
Las formas más habituales de AME son las de tipo I, II y III. En la de tipo I (o AME tipo 1), que es la forma más frecuente de la afección, en la mayoría de los casos habrá dos copias; en la AME tipo II, el escenario más habitual vendrá determinado por tres copias, y así sucesivamente. Con respecto a la supervivencia de los pacientes, la experta aseveró que si no se aplica “un tratamiento específico para la AME, para restituir el gen y la proteína”, la esperanza de vida “será muy limitada” en los cuadros o tipos más graves de la enfermedad y más alta en los casos menos graves.
Difusión: www.farmacosalud.com
2. ¿Qué síntomas o signos clínicos nos deben alertar de la AME?
Si bien la medicación o las terapias actuales para la AME tienen un potencial de acción muy relevante, “la respuesta a las mismas viene determinada por el momento en que empecemos la terapia. Por lo tanto, hay una urgencia diagnóstica, principalmente en los AMEs de tipo I”, advirtió. Se sabe que la enfermedad suele empezar antes de que los profesionales sanitarios observen signos -la degeneración de motoneuronas es previa a tales indicios-, pero como por ahora en España aún no se ha establecido un screening neonatal específico para estos casos, “tenemos que atender a los signos clínicos”, señaló la Dra. Munell. Sea como fuere, es “tan importante hacer el diagnóstico precoz y empezar el tratamiento” lo más precozmente posible, “que una demora de 2-3 semanas puede causar un cambio significativo en la respuesta” terapéutica, avisó la facultativa del Hospital Vall d´Hebron.
Difusión: www.farmacosalud.com
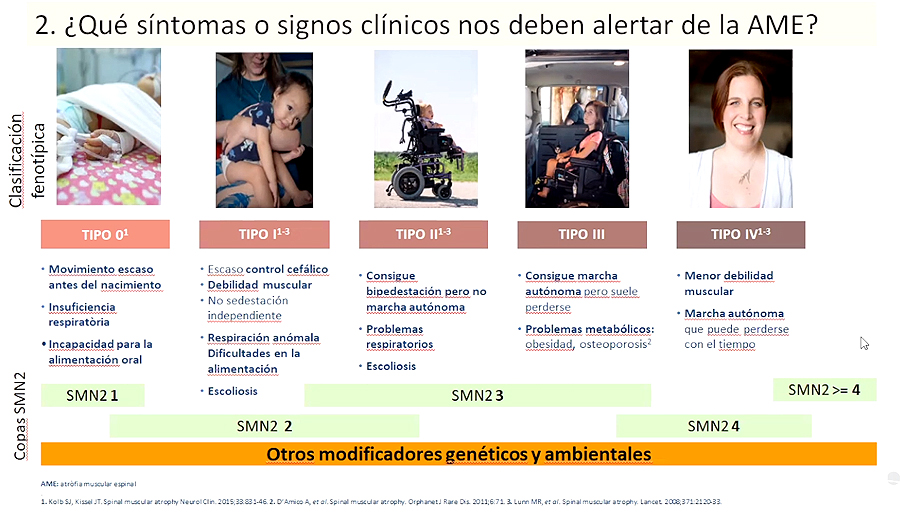
En las personas aquejadas de AME se observan disfunciones motoras y también de índole respiratoria y deglutoria, que son más acusadas en los pacientes que presentan las formas más severas de la patología. Por ejemplo, se necesitará un soporte respiratorio por afectación de la musculatura accesoria respiratoria en aquellos pacientes AME tipo I que no hayan recibido tratamiento específico -y si lo han recibido quizás también, aunque la afectación será más leve-, y probablemente también requerirán ayuda para alimentarse debido a problemas de deglución, no pudiendo nutrirse por vía oral.
Fuente: www.farmacosalud.com
Pacientes con AME tipo I
Las alarmas deben saltar ante un niño hipotónico (no aguanta la cabeza cuando por edad le corresponde poder realizar esa función) que es incapaz de sentarse y que presenta gran debilidad en las extremidades, lo que se conoce como ‘niño en postura de libro abierto’, o sea, no es capaz de elevar las extremidades contra gravedad, o, si lo hace, procede con muy poca fuerza. Este sería un caso típico de AME tipo I.
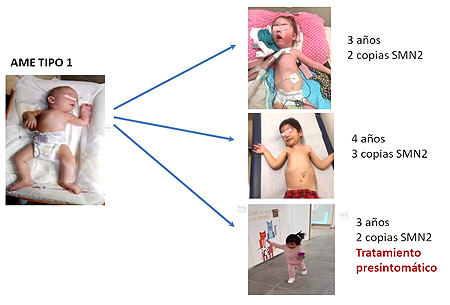
En caso de tener dos copias de SMN2, y si no se aplica un tratamiento que incida en la causa de la enfermedad, al cabo de unos años el paciente estará sobreviviendo gracias a la colocación de una traqueostomía y una gastrostomía, pero no dispondrá de ninguna capacidad motora. Con tres copias, el niño afectado seguramente requerirá ventilación respiratoria nocturna y ayuda para comer, y no podrá sentarse ni caminar.
No obstante, incluso en un AME tipo I con dos copias, si se administra un tratamiento precoz se logran notables progresos, como es el caso concreto de una niña tratada en el centro hospitalario donde trabaja Munell, y cuya imagen se pudo visualizar en una diapositiva (a la izquierda de estas líneas, abajo del todo). Esta paciente es capaz de caminar y de correr y no necesita soporte respiratorio ni ayuda para comer.
A juicio de la especialista, esta niña ejemplifica la enorme relevancia de “distinguir los síntomas de una manera muy precoz” y de “tratar de una manera muy precoz”, o lo que es lo mismo, “empezar el tratamiento lo antes posible” como estrategia para intentar mejorar los resultados terapéuticos.

Signos de alerta de la enfermedad. Hipotonía axial (cabeza y tronco): los pacientes no aguantan la cabeza y, además, se doblan sobre ellos mismos cuando el cuidador intenta sentarlos; hipo/arreflexia: falta de reflejos; pausas durante la lactancia: no pueden respirar y comer a la vez, por lo que hacen pausas; fasciculaciones linguales: temblor en la lengua
Difusión: www.farmacosalud.com
Para ahondar en la ponencia de la Dra. Munell, acceder al vídeo que sigue a continuación:


