Redacción Farmacosalud.com
Pasireotide se ha configurado como una de las alternativas farmacológicas al fracaso terapéutico tras cirugía en casos de acromegalia, una enfermedad asociada en la inmensa mayoría de los casos a un tumor benigno hipofisario que provoca un agrandamiento gradual de distintos tejidos y órganos a lo largo de los años, así como múltiples comorbilidades. Pasireotide es un análogo de la somatostatina de segunda generación (más nuevo) cuya prescripción está limitada a pacientes que, tras cirugía, no han respondido a los análogos de la somatostatina de primera generación, lo que sucede hasta en un 50% de los adultos aquejados de esta patología.
La cirugía transesfenoidal (abordaje del tumor hipofisario por la nariz) es el tratamiento de elección tanto para la acromegalia como para el gigantismo (acromegalia sufrida en edades pediátricas). No obstante, casi el 50% de los pacientes van a necesitar añadir tratamiento médico tras la intervención quirúrgica por persistencia de la afección.

Autor/a: George Hodan
Fuente: www.publicdomainpictures.net
Varias opciones terapéuticas
Por lo tanto, la mitad de esos enfermos van a requerir tratamientos añadidos, para lo cual se dispone de varias opciones:
• En primer lugar, los análogos de la somatostatina de primera generación, que son los fármacos de primera elección. Actúan imitando la acción de la somatostatina, una hormona que inhibe la liberación de la hormona del crecimiento; por lo tanto, son medicamentos que reducen la hormona de crecimiento y tienen, además, efectos a nivel de estabilización o reducción de la lesión hipofisaria.
• Cabergolina: se suele emplear cuando los valores hormonales no son muy elevados o en combinación con los anteriores; tienen la ventaja de ser orales, pero no son tan eficaces.
• También se dispone del antagonista del receptor de GH (hormona de crecimiento) pegvisomant, un fármaco muy eficaz que actúa a nivel periférico impidiendo los efectos nocivos de dicha hormona.
• Finalmente, se puede recurrir a pasireotide, que es un análogo de la somatostatina de segunda generación que tiene una afinidad por los receptores de somatostatina diferente de la de los análogos de somatostatina clásicos. “Actualmente la prescripción de pasireotide, al igual que la de pegvisomant, está limitada a pacientes que no hayan respondido a los análogos de la somatostatina de primera generación, lo que sucede hasta en un 50% de nuestros pacientes adultos. Este fallo terapéutico es aún más frecuente en jóvenes menores de 20 años, ya que el gigantismo en muchas ocasiones está ligado a mutaciones genéticas que pueden hacer al niño/adolescente resistente a los análogos de la somatostatina de primera generación”, expone la Dra. Betina Biagetti, miembro del Área de Neuroendocrinología de la Sociedad Española de Endocrinología y Nutrición (SEEN).
Buscando la personalización de los tratamientos
“Disponer de nuevas opciones terapéuticas siempre es positivo -añade en declaraciones a www.farmacosalud.com-. Sin embargo, los protocolos actuales de tratamiento médico respecto al exceso de hormona de crecimiento son prácticamente iguales para todos los pacientes y todas las edades, igual en la acromegalia que en el gigantismo. En este sentido, desde la SEEN trabajamos activamente para personalizar el abordaje de la acromegalia a través de proyectos como REMAH (Registro Español Molecular de Adenomas Hipofisarios), ACROFAST u otros, pertenecientes al área de conocimiento de Neuroendocrinología, con el fin de mejorar la evidencia en relación a la respuesta terapéutica y tener así un protocolo más personalizado de tratamiento. También colaboramos con las sociedades de pacientes para facilitar información y formación y conocer cuáles son sus verdades prioridades”.
La acromegalia es una enfermedad endocrinológica que, en más de un 95% de los casos, se produce por un tumor benigno hipofisario que secreta hormona de crecimiento de forma continua y no suprimible. Las manifestaciones clínicas más características de esta patología son el incremento del tamaño de los manos y los pies; engrosamiento de la nariz, la boca, la lengua y los labios; cambios en los rasgos faciales y voz ronca; protrusión de la mandíbula- prognatismo-; separación de los dientes -diastema-; dolor de cabeza; hormigueos; dolor de manos -síndrome del túnel carpiano- y en las articulaciones; hiperhidrosis (exceso de sudoración), hipertensión arterial, diabetes, reducción del campo visual, astenia o fatigabilidad, alteraciones del humor o irritabilidad y apnea del sueño.

Dra. Betina Biagetti
Fuente: BERBĒS
“En el adulto con acromegalia, la clínica es larvada”
Tanta diversidad y cantidad de síntomas comporta que se necesiten incluso hasta varios años para alcanzar un diagnóstico. A este respecto, desde la SEEN se alerta que la acromegalia puede llegar a pasar desapercibida, en ocasiones, hasta para los propios profesionales sanitarios. La dificultad en su detección sugiere también que sea complicado estimar el número de aquejados de esta dolencia, cifra que según la Dra. Biagetti se estima en unas “3.000 personas en España”. En cuanto al perfil del paciente, responde a un individuo de entre los 40 y los 50 años, sin predominancia de sexo.
Ante las manifestaciones estéticas de la acromegalia (engrosamiento de la nariz, la boca, la lengua y los labios, etc), uno puede pensar que estos rasgos pueden formar parte de la propia constitución genética de la persona. “En el paciente adulto con acromegalia, la clínica es larvada y se diagnostica tras la aparición de varias comorbilidades, no existiendo una combinación de síntomas que dé un diagnóstico definitivo. Sin embargo, la asociación de ciertas comorbilidades como la hipertensión, diabetes y apnea de sueño de detección reciente, en un paciente que además relata aumento del tamaño de la talla del zapato o anillos y /o cambio en los rasgos faciales, deberían hacer sospechar de la presencia de acromegalia”, indica la experta.
A pesar de que la patología remite en la mayoría de los enfermos, puede persistir mediante la aparición de secuelas físicas y psíquicas, deformidad articular y debilidad muscular, entre otras alteraciones. Sea como fuere, la esperanza de vida para las personas con acromegalia se ha logrado equiparar a la de la población general “gracias al mejor control bioquímico y al abordaje integral de sus comorbilidades”, asegura la Dra. Biagetti a través de un comunicado.
La detección de la enfermedad se retrasa hasta 5-10 años en la edad adulta
Cuando la acromegalia se presenta en la infancia se denomina gigantismo. “A estas edades es frecuente encontrar alteraciones genéticas que pueden ser heredables o no, y es por ello muy relevante realizar estudios genéticos en cualquier persona con gigantismo”, señala la Dra. Biagetti.
La secreción continua de hormona de crecimiento en edades pediátricas tiene la particularidad de aumentar de forma notoria la velocidad de crecimiento, dado que las epífisis (parte de los huesos largos donde se aloja la placa de crecimiento) están aún activas. Su manifestación se caracteriza por una “alta estatura y exceso de crecimiento de las partes acras, los músculos y órganos, lo que resulta en que el niño sea demasiado grande para la edad, y sus rasgos se vuelvan más toscos”, precisa.
Si bien el diagnóstico de la acromegalia resulta esencial para evitar su evolución, en algunos casos -tal y como se ha comentado anteriormente- puede llegar a retrasarse, especialmente en la edad adulta. “En el adulto, como se han cerrado las epífisis, no hay aumento de la estatura y sólo aumentan las partes acras cartilaginosas como son las manos, los pies, orejas, nariz… Debido a que estos cambios físicos son sutiles en el adulto, se tarda unos 5 y 10 años en diagnosticar a estos pacientes. No obstante, en el niño el aumento notorio en la velocidad de crecimiento suele reducir esta brecha”, remarca Biagetti. Otros síntomas en la infancia pueden incluir: pubertad tardía, visión doble o dificultad con la visión lateral (periférica), frente y/o mandíbula prominentes, dolor de cabeza, incremento de la sudoración (hiperhidrosis), menstruaciones irregulares, manos y pies grandes con dedos gruesos, rasgos faciales toscos y debilidad, todo ello vinculado siempre a una elevación en la velocidad de crecimiento.
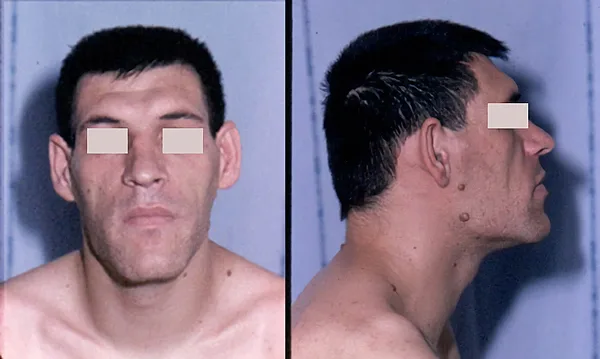
Aspecto facial de un adulto con acromegalia
Autoría y fuente de la imagen: Philippe Chanson and Sylvie Salenave - Acromegaly. Orphanet Journal of Rare Diseases 2008, 3:17. doi:10.1186/1750-1172-3-17
Difusión: Wikipedia
El endocrinólogo está en el centro del manejo de los pacientes de acromegalia, en tanto que, además de confirmar el diagnóstico, coordina las diferentes especialidades para un abordaje multidisciplinar con el cirujano, el oftalmólogo, etc., al tiempo que controla las secuelas e inicia el tratamiento médico cuando es necesario.
Algunos pacientes pueden llegar a necesitar los servicios de salud mental
A juicio de Biagetti, es necesario acompañar al individuo que padece acromegalia no sólo en el momento de la identificación de la patología (momento ideal para asesorarle acerca de las organizaciones de pacientes que existen), sino que, en algunos casos, hay que remitirlo a un especialista en salud mental, ya que el tratamiento y el seguimiento “serán de por vida porque algunas secuelas persisten a pesar de remitir la enfermedad. Por ejemplo, pueden permanecer la artropatía y la debilidad muscular, por lo que el consejo dietético y el ejercicio físico personalizado deben incluirse en el plan terapéutico a largo plazo”.
En esta línea, es fundamental sensibilizar sobre esta afección minoritaria y sus síntomas con el fin de reducir el tiempo del diagnóstico entre los profesionales sanitarios de otras especialidades (dentista, ‘otorrino’, reumatólogo, médico de Atención Primaria) que puedan toparse con las comorbilidades típicas de la acromegalia. También es importante concienciar a la población general. “En un estadio más precoz, el diagnóstico mejora no solamente los resultados de la cirugía, sino también las comorbilidades del paciente”, destaca la endocrinóloga.
Asimismo, las personas con acromegalia pueden sufrir manifestaciones reumatológicas, cardiovasculares, respiratorias, metabólicas, etc. que pueden aumentar la gravedad de la enfermedad: estamos hablando del crecimiento del corazón, tiroides y otros órganos; pólipos en el colon, intolerancia a la glucosa o diabetes mellitus, hipertensión, disminución del deseo sexual, salida de leche por el pezón y patologías coronarias.


