1. Introducción
El término MAT (Microangiopatía Trombótica) es un término patológico usado para describir la enfermedad que cursa con oclusión micro o macrovascular, habitualmente asociada a la formación intraluminal de trombos, y definida clínicamente por la presencia de anemia hemolítica microangiopática y trombocitopenia (MAHAT: microangiopathic hemolytic anemia and thrombocytopenia).

Dra. Lluïsa Hernández Platero
Fuente: Hospital Sant Joan de Déu Barcelona
Debe entenderse como un proceso sistémico, secundario a una lesión endotelial primaria, que conlleva una situación de isquemia en los órganos diana, lo que condicionará la clínica del paciente en función de donde predominen dichos fenómenos isquémicos.
Clínica de la Microangiopatía Trombótica
• Renal
-Hematuria, proteinuria
-Oligoanuria, uremia
-HTA (hipertensión arterial)
• Neurológico
-Irritabilidad, cefalea, somnolencia, confusión, convulsiones, AVC (accidente vascular cerebral), coma
• Cardiovascular
-Isquemia miocárdica, disfunción cardíaca, pericarditis, derrame pericárdico, miocarditis
• Gastrointestinal
-Isquemia intestinal con riesgo de perforación intestinal
-Pancreatitis
-Colestasis, colelitiasis
• Rabdomiólisis
Los principales hallazgos analíticos de la Microangiopatía Trombótica son la trombocitopenia y la anemia hemolítica microangiopática: Hemoglobina < 10 mg/dL; LDH (lactato deshidrogenasa sérica) elevada; descenso haptoglobina; presencia esquistocitos (> 1% es diagnóstico de MAT en ausencia de otra causa conocida); presencia reticulocitos; Test Coombs negativo (excepto en el SHU [Síndrome Hemolítico-Urémico] relacionado con Neumococo o gripe H1N1) e hiperbilirrubinemia de predominio indirecto1.
2. Diagnóstico diferencial
Debido a la evolución rápida y a la severidad que puede presentar la MAT, es necesario establecer un diagnóstico diferencial inmediato desde el punto de vista sindrómico que permita iniciar medidas de soporte en las primeras 24-48 horas de la admisión. Posteriormente se iniciarán las determinaciones necesarias para llegar a un diagnóstico etiológico específico dentro del espectro clínico de Microangiopatía Trombótica.
Autor/a: Rogeriopfm
Fuente: Wikimedia Commons
Microangiopatía Trombótica (MAT) vs Coagulación Intravascular Diseminada (CID)
Se describe CID como el proceso de activación sistémica de la coagulación, secundaria a diversas situaciones clínicas (sepsis, traumatismos o ciertos tumores), que conlleva la aparición de trombosis y sangrado, con frecuente afectación renal.
La diferenciación entre un proceso de MAT y un proceso de CID constituye el principal diagnóstico diferencial a nivel sindrómico que debe realizarse, especialmente en el entorno de Cuidados Intensivos Pediátricos, donde la CID puede ser relativamente frecuente por la tipología de casos que ingresan y su severidad.
El diagnóstico diferencial es importante por la implicación pronóstica y terapéutica que conlleva el diagnóstico de uno u otro proceso.
Como hallazgos clínico-analíticos comunes en ambas entidades se encuentran la anemia, la trombocitopenia, datos de insuficiencia renal, datos de afectación del SNC -Sistema Nervioso Central- (somnolencia, obnubilación, etc.) y/o afectación de otros órganos.
La diferencia fundamental entre un proceso de MAT y CID se basa en la alteración de la coagulación en este último, así como por la presencia de hipertensión arterial que se presenta de forma prácticamente constante en la MAT (la ausencia de HTA debe hacer replantear el diagnóstico).
3. Diagnóstico diferencial etiológico
1. Síndrome Hemolítico Urémico (SHU)
Se describe el Síndrome Hemolítico Urémico (SHU) como la Microangiopatía Trombótica (MAT) de predominio renal. Presenta datos característicos de MAT, como la anemia hemolítica no inmune y la trombocitopenia por formación de microtrombos, y daño renal agudo (AKI), generalmente asociado a HTA.
Se describen diferentes tipos en función del mecanismo etiopatogénico2, siendo importante el diagnóstico diferencial, ya que difiere el abordaje terapéutico:

Escherichia coli (E.coli)
Fuente: COM SALUD (Archivo)
- STEC-SHU, también denominado IA-HUS (Infectious associated- Hemolytic Uremic Syndrome), STEC [VTEC]-SHU o SHU típico
- SHU atípico, que es el SHU mediado por complemento.
1.1. El STEC-SHU es el SHU asociado a infección por bacterias productoras de shiga toxinas, toxinas shiga-like o verotoxinas (STEC = E. Coli productor de Shiga Toxinas). Es la primera causa de SHU en niños. El principal germen implicado es el Escherichia coli, siendo el O157:H7 el serotipo más frecuente. Otras bacterias, como la Shigella dysenteriae, o el Citrobacter freundii, pueden estar también implicadas.
El principal mecanismo fisiopatológico es una lesión endotelial directa producida por la toxina. La unión de la toxina a los receptores celulares Gb3 y Gb4 produce una inhibición de la síntesis proteica condicionando la muerte celular. Esta toxina presenta especial afinidad por las células endoteliales de la microcirculación renal, aunque también por las células de los túbulos proximal y distal, lo que conlleva un doble mecanismo lesional del daño renal agudo. La toxina produce daño también en la mucosa gastrointestinal y en menor medida a nivel de microcirculación del SNC.
Suele presentarse en brotes, en época estival, y clásicamente se presenta tras una fase prodrómica en forma de gastroenteritis con diarrea de inicio brusco, sanguinolenta (70%), dolor abdominal, náuseas, vómitos y fiebre (30%).
1.2 SHU atípico (o SHUa). Constituye el 10% del total de SHU3. Debe considerarse, en general, una enfermedad ultrarrara (siendo la incidencia en Europa de 0,11 casos/millón habitantes).
Se produce por la disregulación de la vía alternativa del sistema de complemento, ya sea por causa genética (mutaciones en los genes que codifican proteínas que participan en la cascada del complemento: factor H [25-30%], MCP [5-10%], factor I, C3, factor B) o adquirida (presencia de anticuerpos frente al factor H).
Cuando se produce la activación del sistema complemento, se forman complejos con actividad C3-convertasa que escinde C3 en C3a y C3b. C3b participa en la creación de MAC (Complejo Ataque a Membrana). En condiciones normales, existen factores reguladores que mantienen bajo control los niveles de C3b (factor H, factor I, MCP). En el SHUa se produce una disregulación de la vía del complemento sobre la superficie celular, lo que condiciona un aumento de C3b y en consecuencia de MAC, lo que produce lesión endotelial y trombosis.
En general, se produce tras una infección o estado inflamatorio que actúa como trigger (desencadenante) de la enfermedad.

Autor/a: PublicDomainPictures
Fuente: Pixabay (free photo)
2. MAT secundarias
Existen otras entidades clínicas que pueden asociarse con el desarrollo de MAT: infecciones (víricas), procesos neoplásicos, fármacos, HTA maligna, TPH (trasplante de progenitores hematopoyéticos), embarazo y posparto, pancreatitis, enfermedades sistémicas autoinmunes o glomerulonefritis.
El origen suele ser multifactorial, por presencia de un estado proinflamatorio, alteraciones del complemento y/o déficit relativo de ADAMTS13.
Tienen especial relevancia en niños, la aciduria metilmalónica, la MAT en contexto de infección por H1N1 y el SHU secundario a infecciones invasivas por serotipos de Streptococcus pneumoniae (pnSHU). En el caso del pnSHU, la bibliografía difiere en considerarla MAT secundaria o IA-HUS (5% de los casos de SHU en niños).
Los pneumococos son productores de la enzima neuraminidasa, que es capaz de exponer el criptoantígeno T en la superficie celular de hematíes y células del endotelio glomerular.
Se produce la unión de anticuerpos naturales al Antígeno T (Ac +Ag), motivo por el cual el Test de Coombs es positivo en estos casos y, finalmente, se produce fenómeno MAT por agregación plaquetaria y lesión endotelial.
La infección por H1N1 presenta mecanismo causal similar al del pnSHU.
3. Púrpura Trombótica Trombocitopénica (PTT)
Se produce a consecuencia de una deficiencia grave de ADAMTS13 (<5-10% actividad). El ADAMTS13 es la enzima plasmática encargada de fragmentar los multímeros ultralargos del Factor de Von Willebrand; el no fraccionamiento de estos multímeros conduce a agregación plaquetaria y lesión endotelial.
La etiología de la PTT puede ser genética: mutación en el gen que codifica para ADAMTS13 o adquirida: presencia de autoanticuerpos frente a ADAMTS13, siendo esta última más frecuente en adultos.
4. Consideraciones en el diagnóstico diferencial
En la actualidad se admite que los signos y síntomas de los diferentes tipos de MAT no son específicos y no permiten realizar un diagnóstico diferencial entre estas entidades. Hasta el 50% de pacientes con PTT presentan disfunción renal y el 50% de SHUa presentan alteraciones neurológicas. En el caso de la SHUa, hasta en un 30% de casos se inician tras una gastroenteritis o presentan diarrea.

Fuente: www.farmacosalud.com / Archivo
Gentileza del Centro Nacional de Microbiología. Instituto de Salud Carlos III de Madrid
Se han descrito casos pediátricos de SHUa, con mutaciones definidas, en los que el desencadenante ha sido una infección STEC.
Sin embargo, la cifra de plaquetas y la severidad de la afectación renal sí que pueden ser orientativas, puesto que, en la PTT, la trombocitopenia es grave (<20.000/mm3) y la afectación renal moderada, y en el SHUa la trombocitopenia es moderada (50.000-100.000/mm3) y la afectación renal grave.
Existen además los denominados overlaping syndromes y las formas incompletas que pueden dificultar el diagnóstico diferencial4.
En términos generales:
-Considerar PTT con anemia hemolítica microangiopática y trombopenia aisladas.
-Considerar SHU ante cuadros de inicio progresivo con anemia subclínica, trombopenia fluctuante y función renal conservada.
-Considerar el diagnóstico de SHUa ante cuadros de HTA severa, con anemia hemolítica microangiopática o trombopenia, aunque no presenten afectación renal.
-Considerar SHUa ante IRC (insuficiencia renal crónica) o progresiva sin anemia hemolítica microangiopática ni trombopenia.
Exploraciones complementarias para el diagnóstico diferencial de la MAT5:
- Historia clínica completa y detallada
- Antecedentes personales y familiares
- Identificación de factores desencadenantes (fármacos, infecciones)
- Exhaustiva exploración física, incluyendo fondo de ojo
- Analítica general habitual de sangre y orina
- Determinación niveles de haptoglobina y LDH
- Determinación niveles de complemento sérico
- Frotis de sangre periférica (esquistocitos)
- Estudio completo coagulación (fibrinógeno, productos degradación del fibrinógeno y Dímero D)
- Investigación infecciones bacterianas (STEC o neumococo) si la clínica es orientativa
- Diagnóstico de enfermedades sistémicas autoinmunes: determinar anticuerpos antinucleares (ANA), anticuerpos anticitoplasma de los neutrófilos (ANCA).
- Diagnóstico de patología infecciosa viral mediante serologías o PCR víricas (CMV -citomegalovirus-, VHC -hepatitis C-, VHB -hepatitis B-, VIH -virus de la inmunodeficiencia humana-, H1N1…)
- Ampliado: T. Coombs, ADAMTS13
5. Manejo médico del MAT/ SHU en el Paciente Crítico
Se trata de pacientes que presentan una clínica aguda con potenciales complicaciones vitales y que precisan de una estricta monitorización para su manejo, motivo por el que se indica su ingreso en UCI (Unidad de Cuidados Intensivos)6.
En UCI se llevará a cabo una monitorización estricta, medidas de soporte y tratamiento dirigido según el diagnóstico diferencial (STEC-HUS -o STEC-SHU-, aHUS -o SHUa-, MAT secundarias).

Fuente: Archivo
Monitorización estricta de:
a. Disfunción renal
-Volemia: peso de los pacientes, presencia de edemas, control del balance hídrico horario.
-Función Renal: realizaremos control analítico seriado del paciente y control estricto de la diuresis mediante sondaje.
-Medio interno: osmolaridad, ionograma con determinación de calcio, magnesio, fosfato y estado ácido-base.
b. Disfunción hematológica
c. Disfunción neurológica: Glasgow, exploración neurológica seriada, realización de EEG (electroencefalografía) y/o TC (tomografía computarizada) según hallazgos clínicos.
d. Problemas gastrointestinales: inspección heces, control estricto de la glicemia. Analítica con perfil hepático completo y enzimas pancreáticas. Control de posibles complicaciones que requieran la valoración de cirugía o realización de TC.
e. Disfunción cardíaca: Determinación de enzimas de lesión isquémica, monitorización ECG (electrocardiograma) y de la TA (tensión arterial) y realización de ecocardiograma a todos los pacientes.
Es posible que, durante el ingreso en UCI, los pacientes requieran la canalización de vías centrales tanto venosas como arteriales. Debemos tener en cuenta el alto riesgo de sangrado debido a la plaquetopenia; se intentará canalizar las mínimas posibles, evitando las arteriales radiales y siempre ecoguiadas por personal experto.
Tratamiento de soporte
1. Medidas de soporte general:
a. Analgesia: convencional, con metamizol y paracetamol, evitando la administración de AINES (antiinflamatorios no esteroideos). Los opiáceos a dosis altas pueden favorecer la aparición de íleo paralítico, empeorando el curso de la enfermedad en el caso de las STEC-SHU.
b. Fluidoterapia: el objetivo es ir hacia un estado de euvolemia en el momento del diagnóstico. Debemos evitar la sobrecarga hídrica, pero corrigiendo la deshidratación en la fase prodrómica de las STEC-SHU, puesto que si no corregimos el componente prerrenal de estos pacientes, empeoraremos su pronóstico de recuperación renal y neurológica.
c. Nutrición: inicio precoz como en cualquier paciente crítico valorando su tolerancia debido a la clínica gastrointestinal y el uso de fórmulas específicas si existe insuficiencia renal establecida.
Autor/a: Thomas Schultz
Fuente: Wikipedia
2. Tratamiento de soporte a nivel7-10:
a) Neurológico: los síntomas pueden estar presentes en un 17-34% de las STEC-HUS y hasta en el 48% de las SHUa.
-Manifestaciones clínicas: irritabilidad, convulsiones, disminución nivel de conciencia, focalidades neurológicas. -Valoración clínica y mediante TC o RM (resonancia magnética).
-Manejo: corrección de la acidosis metabólica, la hiponatremia y la uremia. Tratamientos antiepilépticos con benzodiacepinas levetiracetam o fenitoína. Si Glasgow <8, intubación del paciente y conexión a ventilación mecánica. Estricta vigilancia ante signos de hipertensión intracraneal y, si éstos aparecen, iniciar tratamiento antiedema (SSH, manitol), optimizar sedoanalgesia y valorar el riesgo-beneficio por las posibles complicaciones hemorrágicas de colocar un catéter de presión intracraneal (PIC).
b) Cardíaco:
-Manifestaciones clínicas: disfunción miocárdica, pericarditis, derrame pericárdico o miocarditis que pueden debutar según la severidad del paciente con shock cardiogénico por disfunción ventricular o taponamiento. Isquemia miocárdica con alteración electrocardiográfica y elevación de troponinas. Y por último, arritmias derivadas de las diselectrolitemias.
-Es fundamental la valoración mediante ecocardiografía.
-Manejo: soporte ventilatorio según precise el paciente (Oxigenoterapia, VNI -ventilación mecánica no invasiva-, VM -ventilación mecánica-), control de la hipertensión, corrección de la hiperpotasemia, hipocalcemia, hipomagnesemia y la acidosis. Si hay isquemia, iniciar vasodilatadores coronarios: nitroprusiato.
Si hay disfunción miocárdica, iniciar tratamiento inotrópico con inodilatadores (milrinona) en el caso de asociar hipertensión, o con aminas (dopamina, adrenalina) en el caso de asociar hipotensión; drenaje pericárdico si hay taponamiento que compromete la función del paciente, teniendo en cuenta que se trata de un procedimiento de riesgo por la plaquetopenia del paciente.
c) Gastrointestinal:
-Intestinal: cursan con diarreas, dolor abdominal y distensión que pueden ser la manifestación de fenómenos de isquemia, necrosis y, en las formas más severas, perforación.
-Valorar la necesidad de pruebas radiológicas (ecografía, TC), la valoración por cirugía y la monitorización de la presión intrabdominal.
-Manejo: analgesia, protector gástrico, corrección deshidratación (normovolemia), control de la hipertensión, corrección de la anemia, antibioticoterapia si hay sospecha de perforación.
-Pancreatitis: cursa con dolor abdominal agudo irradiado en cinturón, distensión abdominal, vómitos y elevación de la lipasa y amilasa.
-Realizaremos TC abdominal para su diagnóstico.
-Manejo: analgesia (meperidina), normovolemia del paciente, antibioticoterapia, corrección de la hiperglicemia con insulina, iniciar nutrición, preferentemente enteral, y, si no es posible, parenteral. Valorar inicio de octreotida.
-Diabetes mellitus: se puede presentar en fase aguda debido a los fenómenos de lesión endotelial a nivel pancreático. Se presenta en las formas más severas y se asocia a mayor mortalidad. En más fase tardía como secuela de dichos fenómenos.
-Manejo: su manejo en la fase aguda es mediante infusión continua de insulina.
Autor/a de la imagen: E. Arandes / www.farmacosalud.com
Fuente: Gentileza del Hospital Sagrat Cor de Barcelona (ARCHIVO)
-Colestasis: dolor agudo, ictericia y patrón de colestasis en la analítica. Puede presentarse en fases más tardías.
-Manejo: se deberá realizar tratamiento analgésico y cobertura antibiótica en el caso de alteración de reactantes de fase aguda.
-Hepatitis: cursa con elevación de transaminasas
-Manejo: realizaremos una monitorización estricta y diaria de la función hepática (bilirrubina, coagulación, lactato, nivel de transaminasas), con ajuste o eliminación de aquellos fármacos que se eliminen por metabolismo hepático.
d) Hematológico:
-Anemia: el 80% requerirán transfusiones que estarán indicadas si la (hemoglobina) Hb <7 g/dL o por un descenso de > 2g/dL en 24 horas.
-Plaquetopenia: transfundir si hay hemorragia activa o previo a procedimientos.
-Tratamiento de trombosis: tratamientos antiagregantes, anticoagulantes o fribinolíticos no han demostrado un mejor pronóstico en el curso de la enfermedad y sí un aumento en el riesgo de sangrado y en la mortalidad de estos pacientes, por lo que no están indicados de manera generalizada.
e) Respiratorio:
-Manifestaciones: insuficiencia respiratoria por edema agudo de pulmón o lesiones pulmonares agudas con infiltrados alveolares bilaterales que pueden cumplir criterios de SDRA (síndrome de distrés respiratorio agudo).
-Manejo: corrección de la sobrecarga hídrica. Soporte respiratorio según situación clínica del paciente (oxigenoterapia, VNI, VM). Valorar antibioticoterapia.
f) Otras
-Rabdomiólisis: debilidad, dolor muscular, aumento de CKs (citoquinas) y empeoramiento de la insuficiencia renal. -Manejo: volemia correcta, corrección de acidosis, diuréticos, optimización o inicio de terapia renal de reemplazo.
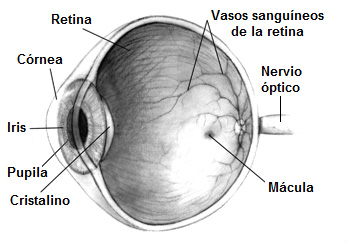
Sección de un ojo humano
Autor/a de la imagen: Modificado de: Human_eye.jpg (14KB, MIME type: image/jpeg) (Wikipedia commons) Human eye cross-sectional view grayscale Copyright: public domain, credit to NIH en:National Eye Institute requested. Original source: http://www.nei.nih.gov/health/macularhole/index.asp. Ojo_humano.jpg: Pixel derivative work: Taty2007 (talk) - Ojo_humano.jpg
Fuente: Wikipedia
-Lesiones oculares: hemorragias retinianas, lesiones isquémicas del nervio óptico. Exploración física y, ante sospecha, realizar fondo de ojo.
g) Renal
-Manifestaciones: insuficiencia renal: acidosis metabólica, diselectrolitemias (hiperpotasemia, hipocalcemia, hiponatremia, hiperfosfatemia), oligoanuria con signos de sobrecarga hídrica y uremia. Hipertensión arterial.
-Manejo: evitar sobrecarga hídrica. Administración de diuréticos de asa (Furosemida). Tratamiento de la hipertensión con ACAs (nicardipino) o centrales (clonidina). Corrección médica de las diselectrolitemias.
-Terapia renal de reemplazo: indicada cuando el tratamiento médico es insuficiente para el control de:
1. Diselectrolitemias
2. Sobrecarga hídrica
3. Uremia
No existe una indicación clara de la técnica de reemplazo renal de elección en estos pacientes.
Clásicamente, en lactantes, se había utilizado la diálisis peritoneal por ser una técnica más sencilla, dada la dificultad en el acceso vascular de los más pequeños, y que no requiere de una formación muy específica para su aplicación. Sin embargo, la técnica elegida dependerá de la experiencia de los equipos y de la situación clínica del paciente. Lo que sí está establecido es que a los pacientes estables se les inicie una terapia intermitente, por asociarse ésta a un mejor pronóstico de recuperación renal en los pacientes con IRA (insuficiencia renal aguda).
Tratamiento dirigido
1) STEC-SHU
a) De soporte: actualmente el pilar fundamental para este síndrome sigue siendo el tratamiento médico de soporte ante las manifestaciones clínicas11.
b) Antibióticos: el uso de antibióticos es controvertido. Su indicación en la fase prodrómica de la enfermedad se asocia a un peor pronóstico, por lo que no está indicado de forma generalizada en aquellos pacientes sin signos de infección sistémica. Sin embargo, si a la hora del diagnóstico el paciente ya ha iniciado antibioticoterapia, se recomienda finalizarla porque su interrupción puede favorecer un empeoramiento del cuadro clínico. En el SHU establecido, ante signos de sepsis y colonización persistente el tratamiento estaría indicado.
c) Pronóstico: la STEC-SHU tiene una mortalidad estimada de entre el 3-5% derivada, principalmente, de las complicaciones neurológicas, cardiovasculares o intestinales. Se estima que hasta el 25% de los pacientes padecerán secuelas por presentar GFR (tasa de filtración glomerular) < 80 mL/min/1,73m2, proteinuria persistente e hipertensión. La anuria de > de 2 semanas se asocia a un peor pronóstico y aquellos pacientes que requieren de reemplazo renal > 4 semanas raramente recuperan función renal.

Autor/a de la imagen: Enric Arandes
Fuente: E. Arandes / www.farmacosalud.com
2) Pn-SHU o SP-HUS (síndrome hemolítico urémico secundario a infecciones invasivas por serotipos de Streptococcus pneumoniae)
a) Antibióticos: inicialmente cefalosporina de 3ª generación + vancomicina; posteriormente desescalar si es posible.
b) Transfusiones: hematíes y plaquetas desplasmatizadas para eliminar anticuerpos contra antígeno TF.
c) Recambio plasmático: terapia de rescate que ha tenido resultados positivos en series cortas de casos sin una evidencia sistemática clara en la mejoría del curso de la enfermedad.
d) Pronóstico: presenta un peor pronóstico con una mortalidad estimada del 10-12%, sobre todo en aquellos casos que asocian meningitis. También presentan peor pronóstico en cuanto a funcionalidad renal, un 23% desarrollará IRC, un 28% proteinuria, y un 19% hipertensión, con mayor porcentaje de trasplante renal.
3) aSHU o SHUa12
a) Ecolizumab: es el tratamiento de primera línea. Anticuerpo monoclonal que se une con gran afinidad a la proteína C5 bloqueando la activación de la vía final del complemento.
b) Recambio plasmático: de rescate como puente hasta el inicio del tratamiento con anticuerpo monoclonal donde no esté disponible.
c) Pronóstico: aunque el ecoluzimab ha atenuado de forma notable el curso de la enfermedad, los pacientes con mutaciones más favorables presentan recaídas y hasta el 25% progresan a un estadio final de enfermedad renal crónica con necesidad de diálisis y trasplante a los 18 años como edad media.
4) MAT secundarias
a) Control de la enfermedad de base o retirada del fármaco.
b) Ecolizumab.
c) Recambio plasmático.
d) Pronóstico: asociado a la enfermedad de base.
Bibliografía
1. Nester CM. Multifaceted hemolytic uremic syndrome in pediatrics. Blood Purif. 2013; 35:86-92.
2. Cavero T, Praga M. Protocolo diagnóstico del síndrome hemolítico-urémico. Medicine. 2015;11(82):4935-7
3. Campistol JM, Arias M, Ariceta G, et al. An update for atypical haemolytic uraemic syndrome: diagnosis and treatment. A consensus document. Nefrologia. 2015;35(5):421-47. doi: 10.1016/j.nefro.2015.07.005. Epub 2015 Oct 9.
4. Scully M, Cataland S, Coppo P, et al; International Working Group for Thrombotic Thrombocytopenic Purpura. Consensus on the standardization of terminology in thrombotic thrombocytopenic púrpura and related thrombotic microangiopathies. J Thromb Haemost. 2017 Feb;15(2):312-322. doi: 10.1111/jth.13571. Epub 2017 Jan 30.
5. Cody E. M, et al. Hemolytic Uremic Syndrome. Pediatric Clinics of North America. 66(1), 235-246. Doi: 10.1016/j.pcl. 2018.09.011.
6. Pérez A, Goñi C. Síndrome hemolítico-urémico. Protocolos de la Sociedad Española de Cuidados Intensivos Pediátricos. https://secip.com/protocolos/
7. Casado J, Loscertales M. Casos clínicos en Patología Infecciosa Pediátrica en UCIP. 2010. Editorial Ergon. ISBN: 978-84-8473-826-8
8. Khalid M, Andreoli S. Extrarenal manifestations of the hemolytic uremic syndrome associated with Shiga toxin-producing Escherichia coli. Pediatr Nephrol. 2018; Nov 1.
9. Fidan k et al. Extra-Renal manifestations of atypical hemolytic uremic syndrome in children.. Pediatr Nephrol. 2018 Aug;33(8):1395-1403
10. Walsh PR, Johnson S. Treatment and management of children with haemolytic uraemic syndrome. Arch Dis Child. 2018 Mar;103(3):285-291.
11. Ardissino G1 et. Early Volume Expansion and Outcomes of Hemolytic Uremic Syndrome. Pediatrics. 2016 Jan;137(1).
12. Chantal Loira et al. An international consensus approach to the managment of atypical hemolytic uremic síndrome in children. Ped Nephrol 2015, Febr. DOI: 10.1007/s00467-015-3076-8.


