Redacción Farmacosalud.com
El Comité de Medicamentos de Uso Humano (CHMP, por sus siglas en inglés) de la Agencia Europea del Medicamento (EMA, por sus siglas en inglés) ha emitido una opinión positiva a KAFTRIO® (ivacaftor/tezacaftor/elexacaftor), en combinación con Kalydeco® (ivacaftor) 150mg para el tratamiento de personas con fibrosis quística (FQ) de 12 años y mayores con al menos una mutación F508del y una mutación de función mínima (F/MF) o dos mutaciones F508del en el gen regulador de conductancia transmembrana de la FQ (CFTR), la mutación causante de la FQ más común.
Si se aprueba, más de 10.000 personas con FQ en Europa de 12 años y mayores, y que tengan una mutación F508del y una mutación de función mínima (F/MF), serían candidatos por primera vez a recibir un tratamiento dirigido a tratar la causa subyacente de la enfermedad. Asimismo, las personas de 12 años y mayores, que tengan dos mutaciones F508del y que actualmente son candidatos para uno de los otros tratamientos de la FQ aprobados por la EMA, también serían candidatos para este nuevo tratamiento combinado. Esta terapia no está disponible en España ya que, todavía, no ha recibido la aprobación de comercialización en Europa por la Comisión Europea. La compañía Vertex quedará a disposición de las autoridades españolas para trabajar sobre el reembolso del medicamento.
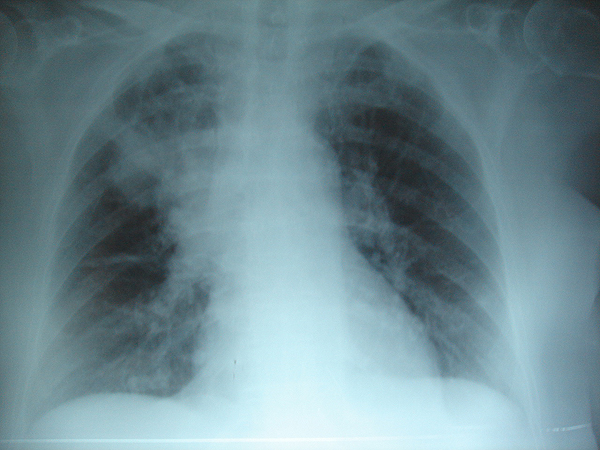
Neumonía apical derecha: en la FQ, la infección crónica da lugar a la destrucción del parénquima pulmonar, ocasionando por último la muerte por insuficiencia respiratoria
Autor/a de la imagen: Joseaperez 17:35, 4 January 2006 (UTC)
Fuente: Wikipedia
Dos estudios internacionales
La opinión positiva del CHMP se basa en los resultados de dos estudios internacionales de fase 3 en personas con FQ: un estudio de 24 semanas en personas de 12 años en adelante con una mutación F508del y una mutación de función mínima y un estudio comparativo directo de cuatro semanas de duración de la terapia de combinación triple en comparación con tezacaftor/ivacaftor en personas de 12 años y mayores con dos mutaciones F508del. Ambos estudios de fase 3 mostraron mejorías estadística y clínicamente significativas en la función pulmonar (porcentaje de volumen espiratorio forzado previsto en un segundo; ppFEV1), que fue la variable principal, y también en todas las variables secundarias clave. En ambos estudios, el régimen combinado de ivacaftor/tezacaftor/elexacaftor en combinación con ivacaftor fue generalmente bien tolerado.
“Los datos clínicos de ivacaftor/tezacaftor/elexacaftor más ivacaftor en personas con fibrosis quística de 12 años y mayores con un genotipo F/F o F/MF no tienen precedentes. Además de las mejoras en la función pulmonar, los datos han mostrado mejoras en múltiples variables de medición de resultados importantes en la FQ, incluida la calidad de vida en personas con FQ de 12 años de edad y mayores con al menos una mutación F508del en el gen CFTR medido por el puntaje del dominio respiratorio (CFQ-R, por sus siglas en inglés)”, declara el Prof. Marcus A. Mall, director médico y jefe del Departamento de Pediatría, División de Neumología, Inmunología y Medicina Intensiva del Centro Médico de la Universidad Charité de Berlín. “Tanto la comunidad médica, como la de pacientes, esperan que más personas con FQ puedan ahora beneficiarse de los moduladores de CFTR”, añade el experto.
Sobre la Fibrosis Quística
La fibrosis quística es una enfermedad rara, genética y degenerativa que afecta a aproximadamente 75.000 personas en todo el mundo. Es una patología multisistémica que empeora progresivamente con el tiempo, afectando a los pulmones, hígado, tracto gastrointestinal, senos nasales, glándulas sudoríparas, páncreas y al aparato reproductor. La FQ está causada por una proteína CFTR (regulador de la conductancia transmembrana de la fibrosis quística) ausente o defectuosa resultante de las mutaciones en el gen CFTR. Para tener FQ, los niños deben heredar dos genes CFTR defectuosos, uno del padre y otro de la madre.
Aunque hay muchas mutaciones conocidas en el gen CFTR que pueden causar la enfermedad, la gran mayoría de las personas con fibrosis quística tienen, al menos, una mutación F508del. Estas mutaciones pueden determinarse mediante una prueba genética o una prueba de genotipado. La función defectuosa o la ausencia de la proteína CFTR dan como resultado un flujo deficiente de sal y agua dentro y fuera de las células en varios órganos. En los pulmones, esto conduce a la acumulación de una mucosidad pegajosa y anormalmente espesa, que puede causar infecciones pulmonares crónicas y daño pulmonar progresivo en muchos pacientes, que finalmente conducen a la muerte. La esperanza media de vida de los pacientes con fibrosis quística se sitúa en torno a los 30 años de edad.


