Redacción Farmacosalud.com
Amiloidosis es un término genérico que hace referencia al depósito extracelular de fibrillas anormales insolubles compuestas por diferentes subunidades de bajo peso molecular (entre 5 y 25 kDa). Estos depósitos proceden de proteínas solubles que, tras sufrir cambios conformacionales, adoptan una estructura predominante de hoja plegada beta alineadas de forma antiparalela1.
Según los órganos afectados, las amiloidosis se pueden clasificar en formas sistémicas (los depósitos se producen en múltiples órganos) o formas localizadas (depósitos circunscritos a un solo órgano o tejido). El término amiloidosis cardiaca hace referencia a la afección cardiaca como consecuencia del depósito amiloideo en el tejido cardiaco, ya sea en el contexto de una afección sistémica (como es más frecuente) o de una forma localizada. Aunque varios tipos de amiloide pueden infiltrar el corazón, sólo la variedad senil, la secundaria (AA), la primaria (AL) y algunas formas de las hereditarias (ATTR, AApoA-I y AFib) pueden producir clínica cardiovascular significativa1.
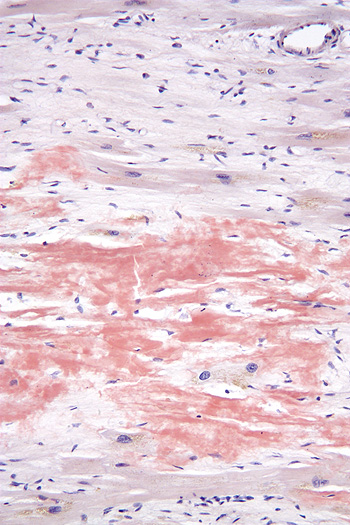
Micrografía revelando material amiloide (mancha rojiza) tras teñir tejido cardíaco con rojo Congo en un caso de amiloidosis cardíaca
Autor/a de la imagen: Nephron
Fuente: Wikipedia
Amiloidosis AL
La amiloidosis de cadena ligera amiloide (AL) es un trastorno hematológico raro y potencialmente mortal que puede afectar a la función de múltiples órganos2,3. Aproximadamente, entre 30.000 y 45.000 pacientes en Estados Unidos y en la Unión Europea tienen amiloidosis AL4.
La amiloidosis AL aparece normalmente por encima de los 50 años, si bien puede aparecer antes. La distribución por sexos es similar o con ligero predominio en los varones, según las series1. La enfermedad se origina cuando la médula ósea produce fragmentos anormales de anticuerpos llamados cadenas ligeras, que se agrupan para formar una sustancia llamada amiloide. Estas agrupaciones de amiloide se depositan en tejidos y órganos vitales e interfieren con la función de órganos normales, causando finalmente deterioro de los órganos2,3. La AL es el tipo más frecuente de amiloidosis. La amiloidosis AL afecta con frecuencia al corazón, los riñones, el tubo digestivo, el hígado y el sistema nervioso y es potencialmente mortal si se deja sin tratar2,3. El diagnóstico a menudo se retrasa y el pronóstico es malo debido a la afectación avanzada multiorgánica, especialmente la cardíaca2,3.
Los pacientes con amiloidosis AL con mayor frecuencia presentan signos y síntomas de cardiopatías o nefropatías5. También son comunes los síntomas de insuficiencia cardíaca, como la disnea de esfuerzo o de reposo y la ortopnea. Éstos pueden estar acompañados de indicios de insuficiencia ventricular derecha como el edema periférico. El paciente puede referir molestias torácicas o dolor torácico, tanto específicas como no específicas del dolor similar al de la angina de pecho5. A todo esto, un paciente puede presentar varios tipos de arritmias. La fibrilación auricular es común. La muerte súbita es un tipo de fallecimiento en pacientes con amiloidosis AL o cualquier tipo de amiloidosis cardíaca que se cree que puede deberse a disociación electromecánica cardíaca o arritmias ventriculares. El síncope y los mareos son manifestaciones comunes y pueden deberse a arritmias o neuropatías autonómicas5.
Menos de un 5% de los pacientes con amiloidosis de cadena ligera cardíaca presentan una afectación cardíaca aislada, y es la presencia de síntomas no cardíacos relacionados la que indica que se trata de una enfermedad sistémica más que de una patología puramente cardíaca5.
Los pacientes con amiloidosis LA suelen tener mal pronóstico, con una mediana de supervivencia estimada que va de seis meses a tres años dependiendo de la población de pacientes y de los datos usados6. Actualmente no hay opciones de terapia aprobadas por organismos reguladores como la Agencia Europea de Medicamentos (EMA) o la Administración de Drogas y Alimentos de los Estados Unidos (FDA, Food and Drug Administration) para tratar esta enfermedad agresiva7,8.
Daratumumab + ciclofosfamida, bortezomib y dexametasona
El estudio de fase 3 aleatorizado ANDROMEDA ha evaluado la formulación subcutánea (SC) de daratumumab en el tratamiento de pacientes con amiloidosis de cadena ligera de nuevo diagnóstico. Los datos demuestran que daratumumab SC en combinación con ciclofosfamida, bortezomib y dexametasona (D CyBorD) alcanza una tasa de respuestas completas hematológicas (RC) significativamente mayor, 53% frente a 18% (P<0,0001), frente a CyBorD[9]. Además, el tratamiento con D-CyBorD retrasa el tiempo hasta el deterioro importante de órganos, la progresión hematológica o la muerte (DIO-SLP) y mejora una supervivencia sin acontecimientos (DIO-SLA) según los criterios DIO-SLP con el tiempo hasta el inicio del siguiente tratamiento9. La combinación muestra un perfil de seguridad consistente con el de daratumumab SC o CyBorD solos.
Autor/a: Marco Verch
Fuente. Flickr / Creative Commons
Los resultados de ANDROMEDA demuestran que el objetivo primario, la tasa de RC hematológica, fue del 53% para D-CyBorD y del 18% para CyBorD (Odds Ratio = 5.1; 95% intervalo de confianza [IC], 3.2-8.2; P <0.0001)9. Además, los pacientes que recibieron D-CyBorD alcanzaron mayores tasas de respuesta hematológica global (92% frente a 77%) y de respuesta parcial muy buena o mayores (≥RPMB; 79% frente a 49%) que los pacientes que recibieron CyBorD. Entre los 195 pacientes que respondieron al tratamiento en el grupo de D-CyBorD, la mediana de tiempo hasta ≥RPMB/RC fue de 17/60 días en comparación con los 193 pacientes del grupo de CyBorD cuya mediana de tiempo hasta ≥RPMB fue de 25/85 días9.
La tasa de respuesta en órgano a los seis meses casi aumentó al doble en los pacientes tratados con D-CyBorD frente a CyBorD, tanto en las respuestas cardíacas (42% frente a 22%; P=0,0029) como renales (54% frente a 27%; P<0,0001)9. Además, la DIO-SLP (cociente de riesgo=0,58; IC del 95%, 0,36-0,93, P=0,0224) y la DIO-SLA (cociente de riesgo=0,40; IC del 95%, 0,28-0,57, P<0,0001) favorecieron el grupo de D-CyBorD, demostrando un retraso significativo del deterioro de órganos importantes, progresión hematológica o muerte, así como mejora de la supervivencia sin acontecimientos. Además, el combo D-CyBorD, que se administra por vía subcutánea, ayuda a limitar la sobrecarga de líquidos intravenosos, un factor importante del tratamiento en el contexto de pacientes con afectación cardíaca9.
Referencias
1. García-Pavía P, Tomé-Esteban MT, Rapezzi C. Amiloidosis. También una enfermedad del corazón. Rev Esp Cardiol. 2011;64(9):797–808
2. Desport E, Bridoux F, Sirac C, Delbes S, Bender S, Fernandez B, Quellard N, Lacombe C, Goujon JM, Lavergne D, Abraham J. Al amyloidosis. Orphanet journal of rare diseases. 2012 Dec;7(1):54.
3. Merlini G, Comenzo RL, Seldin DC, Wechalekar A, Gertz MA. Immunoglobulin light chain amyloidosis. Expert review of hematology. 2014 Feb 1;7(1):143-56.
4. Lousada I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light chain amyloidosis: patient experience survey from the Amyloidosis Research Consortium. Advances in therapy. 2015 Oct 1;32(10):920-8.
5. Mareedu R, Migrino RQ. Guía de la amiloidosis AL (de cadena ligera). Amyloidosis Foundation. 2016. [Internet]. http://www.amyloidosis.org/wp-content/uploads/2016/11/Physicians-Guide-AL-2016-spanishREV.pdf
6. McCausland KL, White MK, Guthrie SD, Quock T, Finkel M, Lousada I, Bayliss MS. Light chain (AL) amyloidosis: the journey to diagnosis. The Patient-Patient-Centered Outcomes Research. 2018 Apr 1;11(2):207-16.
7. Leng S, Bhutani D, Lentzsch S. Amyloid Therapy and Targets. Clinical Lymphoma, Myeloma and Leukemia. 2019 Sep 1;19:S49-52.
8. European Medicines Agency. DARZALEX summary of product characteristics. Available at: https://www.ema.europa.eu/en/documents/product-information/darzalex-epar-product-information_en.pdf Last accessed: June 2020.
9. Kastritis, E. et al. Subcutaneous Daratumumab + Cyclophosphamide, Bortezomib, and Dexamethasone (CyBorD) in Patients with Newly Diagnosed Light Chain (AL) Amyloidosis: Primary Results from the Phase 3 ANDROMEDA Study [LBA]. To be presented at European Hematology Association 2020 Annual Congress.


