Dr. Fernando Santos Rodríguez / Dr. Víctor Navas Serrano
Introducción
La acidosis tubular renal distal (ATRd) es una de las tubulopatías más prevalentes dentro de un grupo de enfermedades minoritarias. La ATRd puede ser hereditaria o adquirida, comunicándose una prevalencia estimada de 0,46 / 10.000 habitantes1, si bien aquellas de origen primario o hereditario tendrían una prevalencia inferior a 1 caso por 100.000 habitantes; por lo que en nuestro país el total de pacientes con diagnóstico de ATRd primaria estaría entre 300 y 4002.
La clínica en los primeros meses de vida (e incluso días) se caracteriza por náuseas, vómitos, cierto grado de deshidratación, polidipsia y poliuria, junto con fallo de ganancia pondero-estatural. En la bioquímica vamos a comprobar la existencia de hipobicarbonatemia, hipercloremia y anión GAP en sangre normal. Adicionalmente suele cursar con hipokalemia. En la orina es característico encontrar la presencia de hipercalciuria, hipocitraturia, pH >5,5 y anión GAP positivo en coexistencia con la acidosis metabólica.

Drs. Fernando Santos Rodríguez (a la izq. de la imagen) y Víctor Navas Serrano
Fuente: Drs. Santos Rodríguez y Navas Serrano
Prácticamente la totalidad de los enfermos diagnosticados en la edad pediátrica van a corresponder con formas hereditarias. En la mayoría de los casos se presenta con un patrón de herencia autosómico recesivo, habiéndose descrito hasta la fecha 6 genes implicados en dicha enfermedad (tabla 1). Los más frecuentes en nuestro medio (más del 80%) son los que codifican para la subunidad V1B1 y V0A4 de la bomba ATPasa de H+, y son concretamente los genes ATP6V1B1 y ATPV0A4. La consecuencia fisiopatológica de la alteración de la bomba ATPasa de H+ es la incapacidad para excretar hidrogeniones a la luz del túbulo, generándose una orina con un pH más alcalino (y una incapacidad de acidificar la orina ante sobrecargas de ácido en la sangre). Otros genes responsables descritos en casos aislados son FOXI1, el WDR72 y el ATP6V1C2, este último descrito recientemente en octubre de 20193.
El tratamiento de estos pacientes consiste en la administración de agentes alcalinizantes de forma crónica. Deben evitarse las sales de sodio, ya que este catión incrementa la hipercalciuria que está presente en la mayoría de los enfermos.
Consecuencias clínicas a largo plazo
El mayor riesgo vital para estos pacientes a lo largo de toda su vida es la aparición de una urgencia metabólica que curse con hipokalemia y acidosis grave. Esta urgencia metabólica puede aparecer a lo largo de la vida en un 25% de los pacientes4, en su mayoría motivada por crisis de hipokalemia, con la consecuente debilidad muscular, parálisis muscular, riesgo de distrés respiratorio, shock, coma y muerte. Se han publicado series de pacientes con una mortalidad del 11% por dicha causa5. En esta emergencia la corrección de la acidosis debe ser realizada antes del tratamiento de la hipokalemia. La administración de suplementos de potasio antes de la compensación de la acidosis no es claramente eficaz.
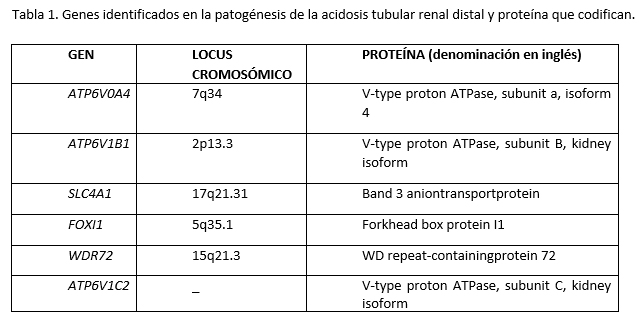
Adaptada de la referencia 6
Se ha estimado que la esperanza de vida de los enfermos con ATRd en el Reino Unido es de 72 años, frente a los 79,4 y 83,1 para varones y mujeres respectivamente en dicho país. Además de la mortalidad atribuida por los episodios de crisis metabólica, esta disminución en la esperanza de vida puede deberse a dos grandes grupos de enfermedades: enfermedad renal y enfermedad ósea (figura 1)6.
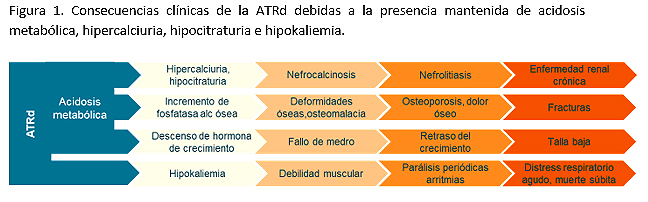
Extraída de la referencia 6
Enfermedad renal
La acidosis metabólica crónica no bien corregida junto con la hipocitraturia, hipercalciuria e hipokalemia son factores de riesgo para la nefrocalcinosis, litiasis renal, aparición de quistes renales y disminución del filtrado glomerular. La nefrocalcinosis, esto es la presencia de calcio en el parénquima renal, es muy prevalente: el 88-94% de los pacientes van a presentarla7,8 y no es reversible con ningún tipo de actuación terapéutica. La litiasis renal se ha descrito en un 12-24%8,9. Teniendo en cuenta que en nuestro medio la litiasis tiene, en la población general, una prevalencia aproximada del 9%, sin duda en la ATRd su presencia puede ser de más del doble que en la población general. Un 82% de los pacientes entre los 20 y 60 años van a presentar una ERC (enfermedad renal crónica) estadio 2 o superior8, y la enfermedad renal crónica estadio 5 se ha descrito en un 2,1-5% de los pacientes10,11. Este aspecto va a tratarse más en detalle en el apartado siguiente, ya que existen varias publicaciones muy recientes a este respecto.
Enfermedad ósea y alteraciones del crecimiento
La acidosis metabólica crónica representa un gran riesgo para la salud ósea. En niños, la presencia de acidosis mantenida, en especial la nocturna, va a dificultar la síntesis y su normal liberación de la hormona de crecimiento, con las consecuencias habituales de talla baja y fallo de medro. En estos pacientes el tratamiento con hormona de crecimiento no está indicado, ya que la normalización del equilibrio ácido-base vendrá seguido de una velocidad de crecimiento adecuada.
La acidosis metabólica va a ser responsable de la enfermedad ósea asociada en estos pacientes: osteoporosis, descrita en un 43% de los niños y en un 90% de los adultos12,10; osteomalacia, reportada entre un 9,6 y 23,3% de los pacientes adultos13,14; deformidades óseas, descritas en un 25% de los niños12; fracturas patológicas notificadas en un 6,3% de estos enfermos10 y retraso del crecimiento y talla baja en 50-79,1% de los niños7.
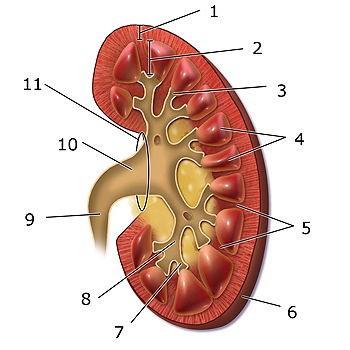
Esquema del riñón: 1. Corteza renal, 2. Médula renal, 3. Papila renal, 4, Pirámide renal, 5. Columna renal, 6. Cápsula fibrosa, 7. cáliz menor, 8. cáliz mayor, 9. Uréter, 10. Pelvis renal, 11. Hilio renal.
Autor/a de la imagen: Modificado de BruceBlaus - Wikimedia Commons file:Blausen 0593 KidneyAnatomy 02.png
Fuente: Wikipedia
Nuevas publicaciones en ERC y ATRd
El hallazgo de nuevos genes responsables de ATRd, la confirmación en cohortes recientes de que existe un significativo porcentaje de casos de ATRd primaria con estudio genético no concluyente, la disponibilidad de un nuevo fármaco para el tratamiento alcalino y el conocimiento del riesgo de insuficiencia renal crónica en la evolución a largo plazo, son todos factores que han propiciado la investigación y publicación de nuevos estudios sobre ATRd.
En relación con el deterioro de la función renal, el análisis de series procedentes de Turquía15 y de España16,17 han confirmado que entre un 28% y 43% de pacientes con formas primarias de ATRd diagnosticadas durante la infancia desarrollan fallo renal crónico, en la edad adulta. Concretamente, en un artículo publicado recientemente con un seguimiento de una cohorte de 16 pacientes con diagnóstico de ATRd genéticamente confirmado, el 43% de los pacientes presentaba un FG <35ml/min/1,73m2 tras un seguimiento de 40 años. La prevalencia de la ERC en esta cohorte fue del 41 y 71% a los 20 y 40 años de seguimiento. En la población general esta prevalencia en nuestro medio es del 5 y 17%. La pérdida de la función renal se estableció en 2 ml/min durante los años 10 y 20 del seguimiento y de 1,65ml/min durante el periodo de los 20 a 40 años17. El mecanismo responsable de este deterioro de la función renal glomerular está aún por dilucidar, habiéndose invocado factores relacionados con el mal control de la acidosis, la presencia de nefrocalcinosis y una talla baja. Serán necesarios estudios dirigidos a esclarecer la causa de la insuficiencia renal en estos enfermos y la mejor forma de prevenir esta complicación.
Otro artículo recién publicado de Bianic et al18 sugiere que la prevalencia de ATRd en el Reino Unido puede estimarse entre un 0,03 y 2,1 por 10.000 individuos, coincidente con la de otras series previamente existentes en la literatura, si bien el número de casos registrados con adecuada codificación diagnóstica es muy inferior. Es, así pues, necesario incrementar el conocimiento de la enfermedad con el fin de conseguir datos más fiables sobre la epidemiología de ATRd, tanto en sus formas primarias como secundarias.
Guías europeas y tratamiento
Recientemente, y en respuesta a una iniciativa de la European Society for Paediatric Nephrology y de la European Network for Rare Kidney Diseases (ERKNet), un grupo de expertos ha elaborado unas recomendaciones actualizadas sobre el tratamiento y control clínico de los pacientes con ATRd, tanto primaria como secundaria, bajo el liderazgo del profesor Detlef Bockenhauer del GOSH de Londres19. Estas guías encuentran su justificación en que la ATRd es una enfermedad rara sobre la que existe muy limitada experiencia en unidades hospitalarias individuales, que es en gran medida desconocida por los nefrólogos de adultos6, y los últimos estudios indican que el tratamiento recibido por los enfermos durante su seguimiento no es totalmente adecuado8. Las recomendaciones principales, que se presentan de forma resumida a continuación, se basaron en los datos aportados por la bibliografía y en el consenso de opiniones de expertos por no existir publicaciones que permitieran aplicar una metodología basada en la evidencia científica.
Referente al diagnóstico:
• En un paciente con síntomas sugerentes de ATRd debe de obtenerse información extensa sobre datos clínicos, bioquímicos y radiológicos que permitan asegurar el diagnóstico.

• Se debe de intentar hacer un estudio genético en todos los casos con sospecha de ATRd primaria. Si éste es negativo, el diagnóstico clínico debería revisarse cuidadosamente y valorar el análisis de otros genes relevantes para el diagnóstico diferencial.
• Debe de realizarse inicialmente y al menos a los 24-30 meses de edad una valoración de la audición en todos los pacientes con ATRd por mutaciones en ATP6V1B1, ATP6V0A4 o FOXI1.
• No se recomienda valoración rutinaria de la mineralización ósea por densitometría o radiología en niños con ATRd. En adultos puede ser útil una densitometría cada 2-3 años para valorar el riesgo de fractura y la adecuación del tratamiento.
• La administración de hormona de crecimiento no está recomendada en niños con ATRd a no ser que persista el hipocrecimiento en presencia de un control metabólico apropiado.
• Se recomienda la valoración de la acidificación urinaria en pacientes con nefrocalcinosis/urolitiasis y concentraciones bajas o límite de bicarbonato en sangre o hipocitraturia con el fin de descartar una ATRd incompleta.
• Debe de valorarse el diagnóstico de ATRd en pacientes adultos con síndrome de Sjögren con urolitiasis o hipopotasemia.
Referente al tratamiento:
• Se recomienda usar suplementos alcalinos para el tratamiento de la ATRd.
• Debe de procurarse mantener valores normales de potasio, cloro y bicarbonato en plasma así como de calciuria.

Autor/a: hue12 photography
Fuente: unsplash.com (free photo)
• Hay diversas formas de suplementar álcali. Desde 2017 se dispone de una preparación microgranular de citrato potásico y bicarbonato potásico de acción prolongada20. Este preparado tiene ventajas relacionadas con una mejor tolerancia y menos frecuencia de dosis, así como una mayor eficacia en el control de la acidosis con respecto a agentes alcalinizantes estándar21. Están pendientes de publicación resultados correspondientes a variables exploratorias del ensayo de extensión de dos años. El tratamiento durante 2 años con el ADV7103 podría traducirse en una ganancia de la densidad mineral ósea en columna vertebral de los pacientes del ensayo, así como una mejora en el z-score de la talla. Sin embargo, son necesarios estudios a largo plazo que confirmen dichos hallazgos en las mencionadas variables clínicas.
• En niños se recomienda una dosis de 2-3 mEq/kg/día de álcali una vez superada la etapa inicial del lactante, en la que pueden precisarse dosis mayores. En adultos, es razonable usar una dosis inicial de 60 mEq/día e ir ajustándola según respuesta.
• Hay que dar suplementos adicionales de potasio si persiste la hipopotasemia con un buen control de la acidosis.
• No se recomienda el uso de tiazidas.
• Los pacientes deben de ser seguidos periódicamente con valoración clínica, bioquímica y ecográfica, en centros con experiencia en la enfermedad.
En resumen, la ATRd es una entidad con repercusión importante sobre la salud de los pacientes pediátricos y adultos, con impacto negativo sobre el crecimiento y la función renal y riesgo de complicaciones agudas graves derivadas fundamentalmente de la hipopotasemia. Estas manifestaciones son en gran medida prevenibles con un correcto diagnóstico y tratamiento de la enfermedad. Así pues, son necesarios estudios dirigidos a extender el conocimiento de la misma (la base genética de las formas congénitas, los mecanismos patogénicos de las formas secundarias y la fisiopatología de las complicaciones) y a procurar un mejor seguimiento de estos enfermos a lo largo de toda su vida en conjunción con un tratamiento que facilite la adherencia y el adecuado control metabólico.
Bibliografía
1. Mumford A, Mumford J, Donald S, Martre C. The Economic impact and variability of managing distal renal tubular acidosis (dRTA) in the UK healthcare setting. Value in Health 2019, 22(3): S914.
2. Santos Rodríguez F. Acidosis Tubular Renal Distal: Introducción, Epidemiología y Genética. En: Santos Rodríguez F, García Nieto VM, Ariceta Iraola G. Actualizaciones en acidosis tubular renal distal. Ed Cyesan; 2019, p. 1-5.
3. Jobst-Schwan T; Verena Klämbt V; Tarsio M; Heneghan JF; Majmundar AJ; Shril s et al. Whole exome sequencing identified ATP6V1C2 as a novel candidate gene for recessive distal renal tubular acidosis. Kidney Int 2019 https://doi.org/10.1016/j.kint.2019.09.026.
4. McSherry E. Renal tubular acidosis in childhood. Kidney international 1981;20:799-809.
5. Sharifian M; Esfandiar N; Mazaheri S; Kariminejad A; Mohkam M; Dalirani R et al. Distal Renal Tubular Acidosis and Its Relationship With Hearing Loss in Children. IJKD 2010; 4:202-6.
6. Torregrosa V; Santos F; González E; Espinosa L; Buades JL; Monteagud-Marrahí E et al. Acidosis tubular renal distal (ATRd): aspectosepidemiológicos, diagnósticos, de seguimiento clínico y terapéuticos. Resultados de una encuesta a un colectivo de nefrólogos. Nefrología 2021: 41(1):2–68.
7. Palazzo V; Provenzano A; Becherucci F; Sansavini G; Mazzinghi B; Orlandini V1. The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney Int 2017; 91, 1243–1255.
8. Lopez Garcia SC; Emma F; Walsh SB; Fila M; Hooman N; Zaniew M et al.Treatment and long-term outcome in primary distal renal tubular acidosis. Nephrol Dial Ttansplant 2019;34: 981-989.
9. Besouw M; Bienias M; Walsh P; Kleta R; van’t Hoff WG; Ashton E et al. Clinical and molecular aspects of distal renal tubular acidosis in children. Pediatr Nephrol 2017; 32:987–996.
10. Zhang C; Ren H; Shen P; Xu Y; Zhang W; Wang W et al. Clinical Evaluation of Chinese Patients with Primary Distal Renal Tubular Acidosis. Intern Med 2015; 54: 725-730.
11. Kamoun T; L. Sfaihi L; Kamoun F; Chabchoub I; Aloulou H; Hachicha M. L’acidosetubulairedistale primitive de l’enfant: etude de 15 observations. La TunisieMedicale 2013; 91:258-262.
12. Caldas A; Broyer M; Dechaux M; Kleinknecht C. Primary distal tubular acidosis in childhood: Clinical study and long-term follow-up of 28 patients. J Pediatr 1992;121:233-241.
13. Jha R; Muthukrishnan J; Shiradhonkar S; Kiran P; Harikumar KVS; Modi KD. Clinical Profile of Distal Renal Tubular Acidosis. Saudi J Kidney Dis Transpl 2011;22(2):261-267.
14. Nilwarangkur S; Nimmannit S; Chaovakul V; Susaengrat W; Ong-aj-Yooth S; Vasuvattakul S et al. Endemic primary distal renal tubular acidosis in Thailand. Q J Med 1990; 74:289-301.
15. Atmis B, Cevizli D, Melek E, Bisgin A, Unal I, Anarat A, Bayazit AK. Evaluation of phenotypic and genotypic features of children with distal kidney tubular acidosis. Pediatric Nephrol 2020; 35:2297-2306.
16. Forero-Delgadillo JM, Gil-Peña H, Alonso-Varela M, Santos F; RenalTubeGroup. Kidney function in patients with primary distal renal tubular acidosis. PediatrNephrol 2021; 36:1931-1935.
17. Gómez-Conde S, García-Castaño A, Aguirre M, Herrero M, Gondra L, García-Pérez N, et al. Molecular aspects and long-term outcome of patients with primary distal renal tubular acidosis. Pediatr Nephrol. 2021 Apr 21. doi: 10.1007/s00467-021-05066-z.
18. Bianic F, Guelfucci F, Robin L, Martre C, Game D, Bockenhauer D. Epidemiology of distal renal tubular acidosis: A study using linked UK primary care and hospital data. Nephron. 2021 Jul:1-10. doi: 10.1159/000516876.
19. Trepiccione F, Walsh SB, Ariceta G, Boyer O, Emma F, Camilla R, et al. Distal Renal Tubular Acidosis: ERKNet/ESPN Clinical Practice Points. Nephrol Dial Transplant 2021 Apr 29.doi: 10.1093/ndt/gfab171.
20. Bertholet-Thomas A, Guittet C, Manso-Silván MA, Joukoff S, Navas-Serrano V, Baudouin V,et al. Safety, efficacy, and acceptability of ADV7103 during 24 months of treatment: an open-label study in pediatric and adult patients with distal renal tubular acidosis. PediatrNephrol 2021; 36:1765-1774.
21. Bertholet-Thomas, A., et al., Efficacy and safety of an innovative prolonged-release combination drug in patients with distal renal tubular acidosis: an open-label comparative trial versus standard of care treatments. Pediatr Nephrol, 2021. 36 (1): p. 83-91.


