INTRODUCCIÓN
El fósforo (símbolo P, número atómico 15) es un elemento químico del grupo de no metales. Tiene una distribución universal en la naturaleza y está presente en la mayoría de los alimentos.
Representa el ~1% del peso corporal y es el segundo mineral en abundancia en el cuerpo humano con una distribución intra y extracelular. Tiene una función como componente estructural del hueso, los dientes y el ADN/ARN, y habilita la polaridad de membranas lipídicas y lipoproteínas circulantes1.
FISIOLOGÍA DEL FÓSFORO
El P depositado en el hueso representa el ~80% del total; es un componente fundamental de la matriz ósea junto con el calcio (Ca), depositándose en su mayor parte en forma de hidroxiapatita. El P extracelular es un 0,1%, con una concentración sérica de P que sufre oscilaciones a lo largo de la vida del individuo, variando desde 7,5 mg/dL en el primer mes de vida hasta 3,5 mg/dL en la edad adulta2,3. Aunque todo el P plasmático se encuentra en forma de anión fosfato, su concentración en plasma se expresa como P elemental. El P extraesquelético supone un ~19,9% y tiene funciones fundamentales para la vida, ya que forma parte de los nucleótidos, de compuestos energéticos como el adenosín trifosfato (ATP) y de fosfoproteínas y enzimas; además, actúa como regulador enzimático e interviene en la fosforilación oxidativa. A nivel intracelular se asienta preferentemente en la mitocondria (cadena respiratoria) y en el núcleo (ácidos nucleicos).
La regulación del P depende sobre todo del umbral renal de la reabsorción del fosfato filtrado que, a su vez, depende de la ingesta de fosfato, de la concentración sérica de éste, y de la actividad de la hormona paratiroidea (PTH), el factor de crecimiento fibroblástico 23 (FGF-23), otras fosfatoninas y el calcitriol. Teniendo claro que el eje FGF-23/alfa-klotho es el principal regulador4:
• La dieta baja o los niveles bajos de P aumentan la síntesis de los cotrasportadores NPT2a en la luz del túbulo contorneado proximal (TCP), aumentando la reabsorción de P (RTP)4.
• Por el contrario, PTH y FGF-23, las dos hormonas fosfatúricas por excelencia, reducen la expresión de dos cotrasportadores sodio-fosfato dependiente, NPT2a y NPT2c, en el TCP, que disminuye la reabsorción de P, aumentando la eliminación y disminuyendo los niveles de P4. La PTH aumenta la actividad de la 1-alfa-hidroxilasa en el TCP para promover la hidroxilación de la vitamina D, aumenta la absorción de Ca y P a nivel intestinal, aumenta la absorción de Ca en los túbulos distales y promueve la eliminación urinaria de P. El FGF-23, producido por los osteocitos y los osteoblastos en respuesta al calcitriol y al aumento de P, Ca y PTH, actúa, al contrario que la PTH, reduciendo la expresión de la 1-alfa-hidroxilasa, que baja los niveles de 1,25 vitamina D. FGF-23 actúa uniéndose en el TCP al receptor FGFR que, a su vez, precisa de un co-receptor, klotho4,5.
La absorción intestinal de P (13 mg/kg/día) es igual a la excreción urinaria de P inorgánico. Así, el recambio de P, aunque solo de 0,1% al día, equivale a renovar diariamente todo el contenido de P inorgánico extracelular. El recambio óseo del P (3 mg/kg/día), debido a la continua remodelación del hueso, es mucho menor6. La reabsorción tubular de P es saturable y su trasporte máximo es solo ligeramente mayor que la carga filtrada usual. Incrementos del nivel plasmático del P producen aumentos paralelos en la excreción renal de P, lo que tiende a mantener el nivel plasmático de P relativamente constante4. Si el aporte dietético de P se mantiene crónicamente elevado, la capacidad máxima de RTP disminuye, contribuyendo a minimizar la elevación del nivel plasmático del P.
El fósforo en la formación del hueso
El proceso inicial de formación ósea comienza con el modelado óseo, la constitución de la forma y el tamaño de las estructuras. En él hay un predominio de hueso cartilaginoso que pronto se somete a remodelado óseo, de modo que el hueso esponjoso cartilaginoso con espículas débiles se convierte en hueso trabecular fuerte. El crecimiento lineal durante la infancia y la adolescencia ocurre por el crecimiento del cartílago en las placas terminales, seguido de la formación de hueso endocondral. El ancho de los huesos aumenta por aposición perióstica7.
Durante la infancia, todos estos procesos se acompañan de reabsorción de la capa ósea interna. El P juega un papel importante en la mineralización, pero también afecta a la proliferación y diferenciación del cartílago, de modo que su déficit produce deformidades óseas, sobre todo cerca de las placas de crecimiento, y retraso estatural7.
RECOMENDACIONES DIETÉTICAS
Existen unas recomendaciones mínimas de aporte de P en la dieta, que van a depender de la edad y del sexo. En los niños de 0 a 6 meses son 100 mg; de 6 meses a un año, 275 mg; de 1 a 3 años, 380 mg; de 4 a 8 años, 405 mg; y de 9 a 18 años, en torno a 1 g. Los requerimientos son más elevados entre la segunda y la cuarta década de la vida (1,5 g y 1,8 g al día), siendo más elevados en el varón1,8. La dosis máxima no está definida, ya que los mecanismos compensatorios son los que van a mantener unos niveles normales, pero se recomienda que no sea superior a 3-4 g al día. Las dietas occidentales, cada vez con mayor ingesta de P por la utilización de los precocinados, asocian mayor riesgo cardiovascular, así como de insuficiencia renal, cáncer y osteoporosis en el adulto joven1.
HIPOFOSFATEMIA
La incidencia de la hipofosfatemia varía dependiendo de la población analizada y de la concentración de P sérico usado para definir hipofosfatemia. Es excepcional que sea carencial, ya que el P se encuentra prácticamente en todos los alimentos. Hasta un 5% de los pacientes hospitalizados pueden tener la concentración de P sérico baja9.
Los valores de normalidad son el principal problema para definir las alteraciones del P, ya que las cifras varían a lo largo de la infancia (Tabla 1)8. La hipofosfatemia grave de menos de 1 mg/dL es rara9.
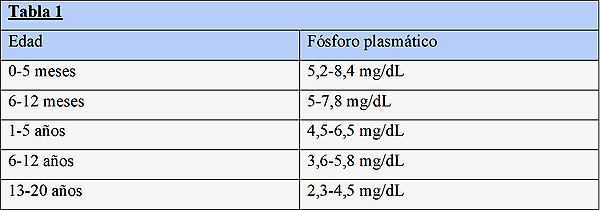
Modificado de Graf L, Reidy K, Kaskel FK. Nutrition Management in Childhood Kidney Disease: An Integrative and Lifecourse Approach. En: Avner ED, et al, eds. Pediatric Nephrology. Springer-Verlag Berlin Heidelberg. 20168.
Hay cuatro mecanismos por los que se llega a la hipofosfatemia9:
• Redistribución de P extracelular al intracelular. El P del espacio extracelular entra en la célula por diferentes factores, entre ellos, debido al aumento de la secreción de insulina o a su administración, la administración de glucagón y epinefrina; o también debido a la realimentación de los pacientes malnutridos, alcohólicos o con anorexia nerviosa. Otras situaciones en las que el P entra en la célula son: la alcalosis respiratoria y el síndrome del hueso hambriento que tiene lugar tras una paratiroidectomía. Al resecar las glándulas paratiroideas, bien por hiperparatiroidismo primario o terciario, o tras una tiroidectomía, existe una osteopenia marcada, lo que hace que se deposite el calcio y el P rápidamente en el periodo postoperatorio inmediato.
• Disminución de la absorción intestinal de P. El P se absorbe principalmente en el intestino delgado; el calcitriol y la vitamina D estimula dicha absorción. La dieta normal contiene el suficiente P para que no haya déficit en ausencia de pérdidas gastrointestinales. Por lo tanto, las carencias debidas a la absorción son el déficit de vitamina D y/o su hidroxilación y todo proceso que impida la absorción de la vitamina D, como la diarrea crónica que inducirá hiperparatiroidismo. Otras causas que impiden la absorción intestinal del fósforo son los antiácidos ricos en aluminio y magnesio. El ayuno total no causa hipofosfatemia, debido a la falta de insulina.
• Aumento de la excreción urinaria de P. El riñón ejerce la mayor influencia en el balance del fosfato. El trasporte renal de fosfato ocurre en el TCP el 60-70% y en el túbulo distal el 10-15% de la carga filtrada. La reabsorción de fosfato se liga a la reabsorción de sodio vía los cotrasportadores sodio-fosfato en la membrana luminal. Estos trasportadores usan el gradiente de concentración de sodio para producir una reabsorción activa del fosfato (en el interior la concentración de sodio es 25 mEq/L y en la luz tubular 145 mEq/L). Los reguladores fisiológicos de la RTP incluyen:
ο La concentración de fosfato: una depleción moderada estimula la reabsorción.
o La PTH, que aumenta la eliminación de P al disminuir los cotrasportadores.
o Fosfatoninas como el FGF-23, el factor de crecimiento fibroblástico 7 (FGF7) y la matriz extracelular fosfoglicoproteína (MEPE), que disminuyen la reabsorción por los cotrasportadores sodio-fosfato.
o La diuresis osmótica por glucosuria en la cetoacidosis diabética o en la hiperglucemia no cetósica, que provoca la pérdida de P.
o Sensores intestinales u óseos que podrían aumentar la excreción de P, aunque no se ha comprobado.
• Técnicas de depuración extrarenal. La hipofosfatemia puede aparecer en pacientes sometidos a terapia renal sustitutiva si la prescripción de diálisis es muy elevada, con eventual necesidad de suplementar con P.
Enfermedades con déficit de fósforo
La hipofosfatemia per se va a ser capaz de aumentar la RTP; si la situación persiste es por presencia de un factor circulante que promueve la pérdida urinaria de P. Puede ser dependiente o independiente de FGF-23.
Dependientes de FGF-23
Son un grupo de enfermedades graves en el que se encuentran los raquitismos hipofosfatémicos hereditarios, que se caracterizan por un problema primario: la pérdida renal de fósforo. En este grupo de enfermedades se incluyen el raquitismo hipofosfatémico ligado al X, el raquitismo hipofosfatémico autosómico dominante, el raquitismo hipofosfatémico autosómico recesivo, la osteomalacia inducida por tumores y una miscelánea.
El raquitismo hipofosfatémico ligado al X (XLH) es la alteración más frecuente. Tiene un defecto en el trasportador del túbulo proximal de fosfato debido a la mutación en el gen PHEX10,11. El gen codifica la endopeptidasa que indirectamente altera la degradación y la producción de FGF-23, la fosfatonina que promueve la excreción urinaria de P y suprime la síntesis de calcitriol; los pacientes con esta alteración tienen raquitismo, deformidades óseas, alteraciones en la dentina, retraso en el crecimiento y también se ha descrito sordera asociada12.
El raquitismo hipofostatémico autosómico dominante tiene pérdida de fosfato con raquitismo u osteomalacia y es el resultado de la mutación en el gen FGF-23 en el cromosoma 12p13, que se transmite de forma autosómica dominante. Esta mutación de FGF-23 es resistente a la escisión de la proteasa, pero conserva sus propiedades fosfatúricas9.
El raquitismo hipofosfatémico autosómico recesivo afecta a los individuos en la infancia tardía y las manifestaciones clínicas y bioquímicas son similares a las del XLH con raquitismo y osteomalacia, aunque algunos pacientes desarrollan osteoesclerosis y sobrecrecimiento óseo. Se han descrito 3 tipos de acuerdo con las diferentes mutaciones detectadas3,9,11:
♦ ARHR1: inactivación del gen DMP1, que codifica la proteína de la matriz de la dentina.
♦ ARHR2: inactivación del gen ENPP1, que codifica la ectonucleótido pirofosfatasa/fosfodiesterasa 1, enzima crítica para la generación del inhibidor de la mineralización, por lo que van a aparecer calcificaciones arteriales generalizadas en la infancia.
♦ AHRH3: con mutaciones en FAM20C, que codifica una proteína-kinasa que fosforila el FGF-23.
La osteomalacia inducida por tumor es similar al raquitismo XLH; si son adultos tendrán osteomalacia. Estos pacientes generalmente tienen tumores de origen mesenquimal, a menudo del tipo hemangiopericitoma esclerosante, que produce hormonas fosfatúricas (FGF-23 y MEPE)9,13.
Otros cuadros más infrecuentes con aumento de FGF-23 son la displasia ósea fibrosa, el síndrome de nevus epidérmico o el síndrome de McCune-Albright, resultado de mutaciones de la subunidad alfa de la proteína G estimuladora9. La hipofosfatemia es una complicación frecuente del trasplante renal y puede ser debida, en parte, al hiperparatiroidismo terciario con niveles elevados de FGF-23. La insuficiencia renal puede contribuir a esta complicación. La administración de diferentes formas de hierro intravenoso produce hipofosfatemia porque contienen restos de carbohidratos que aumentan los niveles circulantes de FGF-23 y con ello la excreción de fósforo9.
Independientes de FGF-23
En este grupo encontramos los síndromes con hipofosfatemia e hipercalciuria, el síndrome de Fanconi y un grupo heterogéneo de trastornos.
La hipofosfatemia e hipercalciuria se describen en el raquitismo hereditario hipofosfatémico con hipercalciuria, que es el resultado de una mutación del gen SLC34A3 en el cromosoma 9q34, que codifica el cotrasportador sodio-fosfato 2c. La mayoría de los pacientes presentan raquitismo y/u osteomalacia con hipofosfatemia, talla baja e hipercalciuria absortiva. En estos pacientes, el calcitriol está normal o elevado y la hipercalciuria viene causada por la absorción intestinal aumentada11,14.
La enfermedad de Dent es una enfermedad recesiva ligada al X cuyo defecto primario se encuentra en el TCP con hipercalciuria, nefrocalcinosis, litiasis, enfermedad renal crónica, raquitismo y proteinuria de bajo peso molecular junto a glucosuria, aminoaciduria y fosfaturia, pero sin bicarbonaturia. Se debe a una mutación en el gen CLCN5 que codifica el trasportador de cloro dependiente de voltaje en el 60% y a una mutación en el gen OCRL1 en otro 15%14.
La hipercalciuria idiopática es un factor de riesgo para la litiasis renal y asocia hipofosfatemia moderada con niveles elevados de calcitriol. Puede presentar defectos tubulares proximales14.
El síndrome de Fanconi es una alteración generalizada de la función tubular proximal con pérdida de todos los compuestos que se reabsorben en el TCP. La consecuencia es: hipofosfatemia, glucosuria, hipouricemia, aminoaciduria y acidosis tubular proximal debido a la pérdida urinaria de bicarbonato. Las concentraciones de calcitriol son bajas o inapropiadamente normales. En niños, la cistinosis, la enfermedad de Wilson y la intolerancia hereditaria a la fructosa son las causas más frecuentes de este síndrome. En adultos es raro y se asocia con mieloma múltiple o con medicaciones9.
Otras causas que pueden aumentar la excreción de fósforo son: diuresis osmótica (la mayoría de las veces debido a glucosuria), diuréticos que actúan proximalmente y los que inhiben la anhidrasa carbónica y la expansión aguda de volumen que disminuye la RTP9.
HIPERFOSFATEMIA
Consideramos hiperfosfatemia cuando los niveles de P están por encima del valor de normalidad. El diagnóstico diferencial es con la pseudohiperfosfatemia, que es la falsa elevación del P por los métodos analíticos y que se produce debido a hipergammaglobulinemia, hiperlipemia, hemólisis, hiperbilirrubinemia, tratamiento con anfotericina B, activador del plasminógeno tisular y heparina15.
La excreción renal de P es tan eficaz en sujetos normales que el balance se mantiene con un mínimo aumento en el P sanguíneo si la ingesta se aumenta a más de 4.000 mg/día. La hiperfosfatemia disminuye la RTP vía supresión de los cotrasportadores sodio-fosfato en la membrana luminal; la PTH aumenta la excreción de P disminuyendo la actividad de los cotrasportadores sodio-fosfato y las fosfatoninas, como FGF-23 y sFRP-4, y aumenta así mismo la excreción de P al suprimir la expresión luminal de los cotrasportadores sodio-fosfato15.
Las causas de hiperfosforemia son las siguientes15:
• Sobrecarga aguda de fosfato (exógena o endógena). Una carga muy elevada de P podría saturar la capacidad renal de excreción.
Una carga endógena se debería a la salida del P al espacio extracelular ya que el P es el mayor anión intracelular; cualquier destrucción de tejidos va a aumentar el P del espacio extracelular. La hiperfosfatemia puede inducir hipocalcemia por depósito tisular de los complejos de fosfato cálcico. Algunos ejemplos son el síndrome de lisis tumoral, la necrosis muscular (rabdomiólisis) y, más raro, la hemólisis o la trasfusión de sangre almacenada. La acidosis láctica y la cetoacidosis diabética (o hiperglucemia grave sola) pueden también aumentar la movilización del fosfato hacia el espacio extracelular.
Una carga exógena de fosfato puede resultar de la ingesta de grandes cantidades de laxantes que contengan fósforo. La contracción de volumen por diarrea o insuficiencia renal leve puede contribuir a hiperfosfatemia15.
• Enfermedad renal aguda o crónica. El P filtrado es aproximadamente de 8 g al día. Si la tasa de filtrado glomerular es de 120 mL/min y la concentración de P es de 4 mg/dL entonces el P filtrado será de 7,2 g/día. Solo del 5 al 20% del P filtrado se excreta normalmente, siendo reabsorbido en su mayor parte en el TCP. Una reducción aguda o crónica en el filtrado glomerular disminuirá la cantidad de P filtrado y excretado; inicialmente se mantendrá disminuyendo la RTP influido por la secreción de PTH y FGF-23. Una vez que el filtrado cae por debajo de 25 mL/min, sin embargo, la RTP se ha suprimido al máximo y no puede mantener el P, por lo que en este momento aparece la hiperfosfatemia que incrementaría la cantidad de P que se filtra y mantendría el balance de P15.
• Aumento primario de la RTP con disminución de la excreción de fosfato. Esto ocurre en pacientes con función renal normal pero que tienen las siguientes alteraciones15:
o Hipoparatiroidismo, bien por deficiencia en la secreción de PTH o por resistencia renal a su efecto (pseudohipoparatiroidismo). Estos pacientes también tienen hipocalcemia por disminución de la reabsorción ósea y por pérdidas urinarias de calcio. Parece que el tratamiento con calcio y vitamina D aumenta la concentración de Ca sérico, que directamente disminuye la RTP. El calcitriol estimula la producción de FGF-23 en el hueso, que puede llegar a suprimir la RTP.
o Acromegalia. Se produce directamente por la estimulación de la RTP por las hormonas de crecimiento o el factor 1 de crecimiento insulin-like.
o Calcinosis tumoral familiar. Enfermedad rara autosómica recesiva con hiperfosfatemia por incremento en la RTP, asociado a elevación del calcitriol. Hay varias mutaciones asociadas, la primera descrita en el gen GALNT3, que codifica la glicosiltrasferasa y que previene la degradación del FGF-23. Posteriormente se han descrito mutaciones en FGF-23 y klotho (el correceptor de FGF-23). Estas mutaciones inactivantes de los genes GALNT3 o FGF-23 podrían conducir a deficiencia de FGF-23 intacto circulante, que es un promotor de la excreción renal de fosfato y que, junto a una deficiencia de klotho, podría producir una resistencia en el órgano final al FGF-23. La calcinosis tumoral familiar es la imagen en espejo de los raquitismos hipofosfatémicos ligado al X y autosómico dominante, ambos con aumento de la actividad de FGF-23. En estos pacientes, el calcio y la PTH están en rango normal. La combinación de hiperfosfatemia y normocalcemia da un producto Ca-P elevado que permite los depósitos en tejido celular subcutáneo y piel.
o Bifosfonatos, vía estimulación de la RTP.
o Intoxicación por vitamina D. Aumenta la absorción intestinal de P y Ca; la elevación del Ca disminuye la eliminación urinaria de P, inhibiendo la secreción de PTH.
USO CLÍNICO
El uso clínico del P está limitado a las situaciones descritas anteriormente, como los trastornos hereditarios del metabolismo del P o las deficiencias por pérdidas de otro origen o desnutrición grave1.
INVESTIGACIONES RECIENTES
Se está trabajando en la relación entre los aditivos que contienen P y el riesgo de enfermedades crónicas en individuos sanos. En animales de experimentación, se ha visto que dietas ricas en P se relacionan con cáncer de pulmón, vejiga y piel. No se alcanza de momento a saber por qué sucede esto, pero lo que es cierto es que el fósforo tiene un papel importante en el correcto funcionamiento del organismo1.
CONCLUSIONES
-El fósforo es un elemento químico imprescindible para el correcto funcionamiento de cuerpo humano y, concretamente, para la correcta formación del hueso.
-Los niveles de fósforo plasmático se mantienen estables en un estrecho margen gracias a los mecanismos de control en el túbulo contorneado proximal.
-Los niveles de fósforo varían a lo largo de la vida y es esencial su determinación ajustada al rango etario de normalidad, especialmente en población pediátrica, ya que su alteración es clave para el diagnóstico de muchas enfermedades óseas.
-La hipofosfatemia se produce principalmente por alteraciones en los genes que regulan los trasportadores de fósforo a nivel renal. Las manifestaciones clínicas principales son deformidades óseas y alteración del crecimiento.
-La hiperfosfatemia se asocia con alteraciones en los genes que regulan los trasportadores de fósforo, pero también se produce por alteraciones en la PTH y vitamina D y en las situaciones en la que el filtrado glomerular se encuentra disminuido.
Bibliografía
1. Calvo MS, Lamberg-Allardt CJ. Phosphorus. Adv Nutr. 2015;6(6):860-2.
2. Minisola S, Peacock M, Fukumoto S, Cipriani C, Pepe J, Tella SH, et al. Tumour-induced osteomalacia. Nat Rev Dis Primers. 2017;3:17044.
3. Santos F, Fuente R, Mejia N, Mantecon L, Gil-Pena H, Ordonez FA. Hypophosphatemia and growth. Pediatr Nephrol. 2013;28(4):595-603.
4. Hogan J, Goldfarb S. Regulation of calcium and phosphate balance. Sterns R. & Lam AQ (eds). UpToDate. 2019. Disponible en: https://www.uptodate.com/contents/regulation-of-calcium-and-phosphate-balance
5. Olauson H, Vervloet MG, Cozzolino M, Massy ZA, Urena Torres P, Larsson TE. New insights into the FGF23-Klotho axis. Semin Nephrol. 2014;34(6):586-97.
6. Berndt T, Kumar R. Novel mechanisms in the regulation of phosphorus homeostasis. Physiology (Bethesda). 2009;24:17-25.
7. Manolagas SC. Normal skeletal development and regulation of bone formation and resorption. Drezner MK & Mulder JE (eds) UpToDate. 2020. Disponible en: https://www.uptodate.com/contents/normal-skeletal-development-and-regulation-of-bone-formation-and-resorption
8. Graf L, Reidy K, Kaskel FK. Nutrition Management in Childhood Kidney Disease: An Integrative and Lifecourse Approach. En: Avner ED, et al, eds. Pediatric Nephrology. Springer-Verlag Berlin Heidelberg. 2016.
9. Yu ASL, Stubbs JR. Hypophosphatemia: Causes of hypophosphatemia. Goldfarb S& Lam AQ (eds). UpToDate. 2020. Disponible en: https://www.uptodate.com/contents/hypophosphatemia-causes-of-hypophosphatemia
10. Fuente R, Gil-Pena H, Claramunt-Taberner D, Hernandez O, Fernandez-Iglesias A, Alonso-Duran L, et al. X-linked hypophosphatemia and growth. Rev Endocr Metab Disord. 2017;18(1):107-15.
11. Robinson ME, AlQuorain H, Murshed M, Rauch F. Mineralized tissues in hypophosphatemic rickets. Pediatr Nephrol. 2019;doi:10.1007/s00467-019-04290-y.
12. Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. 2019;15(7):435-55.
13. Florenzano P, Cipriani C, Roszko KL, Fukumoto S, Collins MT, Minisola S, et al. Approach to patients with hypophosphataemia. Lancet Diabetes Endocrinol. 2020;8(2):163-74.
14. Scheinman SJ, Carpenter T, Drezner MK. Hereditary hypophosphatemic rickets and tumor-induced osteomalacia. Sterns R., Geffner ME & Hppin AG (eds). UpToDate. 2019. Disponible en: https://www.uptodate.com/contents/hereditary-hypophosphatemic-rickets-and-tumor-induced-osteomalacia
15. Stubbs JR, Yu ASL. Overview of the causes and treatment of hyperphosphatemia. Goldfarb S & Lam AQ (eds). UpToDate. 2020. Disponible en: https://www.uptodate.com/contents/overview-of-the-causes-and-treatment-of-hyperphosphatemia
KKI/ES/XLH/0100![]()


