1. ¿Qué es la Deficiencia de Lipasa Ácida Lisosomal (LALD)?

Doctor Emili Ros
Fuente: Hospital Clínic de Barcelona
La deficiencia de LAL es una enfermedad rara (EERR) debida a una mutación en homozigosidad (o heterozigosidad compuesta) en el gen LIPA, que codifica para la proteína “lipasa ácida lisosomal”, una enzima que hidroliza los ésteres de colesterol y triglicéridos captados por las células de las LDL y transportadas a los lisosomas.
Las EERR son trastornos de causa hereditaria que afectan a menos de 1 de cada 2.000 nacidos vivos. Se trata en general de enfermedades graves e invalidantes, de inicio generalmente en la infancia, que cursan con una elevada morbi-mortalidad y suelen ser infradiagnosticadas.
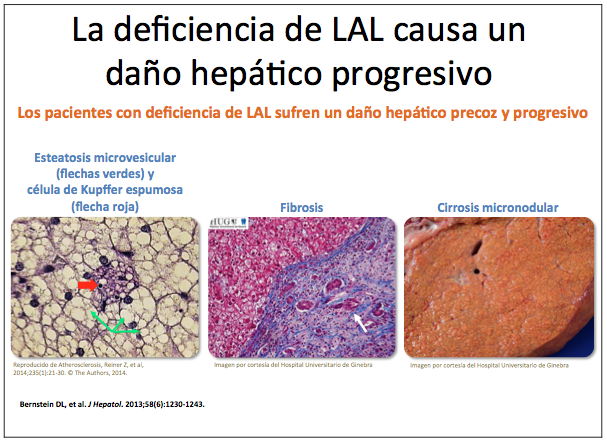
La deficiencia de LAL causa predominantemente enfermedad hepática grave por depósito masivo de ésteres de colesterol, de inicio en general en edad pediátrica. Según el grado de deficiencia de LAL causado por el defecto genético (ausencia o actividad residual), hay dos presentaciones de la LALD: la enfermedad de Wolman, que cursa con hepatoesplenomegalia masiva, malabsorción, calcificación adrenal, caquexia y muerte en el primer año de vida y la enfermedad por depósito de ésteres de colesterol (“cholesterol ester storage disease” - CESD), que tiene un curso clínico muy variable, pudiendo debutar en la infancia o en la edad adulta; esta forma causa hepatomegalia debida a esteatosis masiva de tipo microvesicular y cursa con hipertransaminasemia e hipercolesterolemia grave, que suele estar asociada a hipertrigliceridemia moderada y colesterol HDL bajo. Esta forma de LALD presenta con frecuencia fibrosis hepática que puede progresar a cirrosis franca.
La enfermedad de Wolman recibe el nombre de un patólogo israelí, Moshe Wolman, que describió la primera familia afectada a mediados del siglo pasado. Unos años más tarde Infante en Francia y Fredrickson en EEUU describieron la forma más leve, que quedó etiquetada como CESD. Ambas son formas de presentación de una misma entidad, LALD.
2. ¿Cómo se transmite LALD?
La LALD se transmite mediante herencia autosómica recesiva, lo que implica que ambos progenitores deben ser portadores de un defecto genético funcional en el gen LIPA. Los portadores están asintomáticos, si bien se ha descrito que pueden tener una hipercolesterolemia moderada.
3. ¿Cuál es la prevalencia estimada?
La prevalencia estimada de la LALD en población occidental oscila entre 1:60.000 y 1:130.000. Esto significa que en España debería haber de 350 a 750 casos, de los cuales menos del 10% estarían diagnosticados.
4. LALD es una dislipemia genética. ¿Dónde encaja en la clasificación de dislipemias genéticas?

La dislipemia de la LALD presenta un perfil lipídico similar al de la HF, si bien en la HF los triglicéridos suelen ser normales y el colesterol HDL no está tan bajo. Una diferencia fundamental es que la HF se transmite por herencia autosómica dominante (todos los heterocigotos presentan la alteración lipídica), mientras que la LALD tiene una herencia autosómica recesiva (solo los homocigotos o heterocigotos compuestos manifiestan la enfermedad).
5. ¿Cuál es el mecanismo fisiopatológico asociado a LALD, y qué signos y síntomas a nivel lipídico esperaríamos encontrar?
El mecanismo fisiopatológico de la LALD depende del déficit de actividad LAL, que conlleva el acúmulo celular masivo (predominantemente hepático) de ésteres de colesterol y, en menor medida, triglicéridos.
La lipidosis hepática causa hepatomegalia y elevación de transaminasas e induce fibrosis, que con frecuencia progresa a cirrosis, con todas sus consecuencias (fallo hepático, hipertensión portal, hemorragia por varices esofágicas o gástricas, ascitis, etc.).
La retención lisosomal de colesterol esterificado conduce a una disminución del colesterol libre intracelular, lo cual estimula la síntesis del esteroide y la secreción hepática de VLDL y apoB, con el consiguiente aumento de los triglicéridos y el colesterol LDL circulantes. El colesterol HDL bajo se explica porque la deficiencia intracelular de colesterol libre limita la regulación al alza de la proteína ABCA1, que transporta colesterol a la ApoA-I para formar las HDL nacientes.
6. ¿Puede la dislipemia poner en riesgo otros órganos y/o sistemas y si es así cuáles?
Además de al hígado, la deficiencia de LAL puede afectar a múltiples órganos y sistemas, incluyendo el cardiovascular, el digestivo y el bazo. La dislipemia de la LALD puede tener el mismo efecto aterogénico que otras hipercolesterolemias graves, conduciendo a aterosclerosis prematura y sus complicaciones clínicas (infarto de miocardio y accidente vascular cerebral). Sin embargo, el hecho de que la LALD conduce con cierta frecuencia a fallo hepático, con la consiguiente reducción del colesterol y necesidad de trasplante hepático, hace que las complicaciones vasculares de esta enfermedad sean mucho menos llamativas que las hepáticas.
Fuente: Archivo
7. ¿Cómo se diagnostica LALD y qué otras patologías debemos tener en cuenta en el Dx diferencial de LALD?
El diagnóstico de la LALD se basa en la demostración de una baja actividad LAL en sangre periférica mediante un simple test en gota seca. Es conveniente confirmar el diagnóstico genético mediante secuenciación del gen LIPA.
Es evidente que para sospechar LALD hay que conocer la existencia de esta enfermedad y sus características clínicas. Si no, bastantes pacientes con LALD no diagnosticada estarán “escondidos” entre casos de hepatopatía crónica de causa desconocida y cirrosis criptogénica o de hipercolesterolemia tipo HF sin diagnóstico genético. El hecho de que es una enfermedad que progresa lentamente a hepatopatía crónica avanzada obliga a hacer el diagnóstico cuánto antes porque actualmente hay un tratamiento, la sebelipasa alfa, que tiene muchas posibilidades de detener esta progresión. Los hepatólogos pediátricos y los lipidólogos de niños y adultos son los especialistas que deberían sospechar y diagnosticar la LALD.
El diagnóstico diferencial hepatológico de la CESD debe hacerse en niños y jóvenes que presentan hepatomegalia,hipertransaminasemia e hígado graso junto con hiperlipemia, una vez descartadas las causas comunes de hepatopatía infantil, fundamentalmente vírica y metabólica.
La biopsia hepática demostrando esteatosis microvesicular con afectación de células de Kupffer debe hacer sospechar el diagnóstico, que se confirmará mediante el test de actividad LAL en gota seca.
Para el diagnóstico diferencial de la CESD en la clínica de lípidos deben considerarse los niños y adultos jóvenes con un fenotipo lipídico de hipercolesterolemia primaria compatible con HF, pero sin diagnóstico genético (ausencia de mutaciones funcionales en los genes LDLR, APOB, PCSK9 y ARH), con cHDL bajo y un fenotipo lipídico poco llamativo en los padres, si bien puede haber hermanos afectos. La presencia de hepatomegalia, alteración de las transaminasas e hígado graso por ecografía en ausencia de obesidad deben hacer sospechar el diagnóstico y conducir a la realización de un test de LAL en gota seca.
8. ¿Qué manejo y recomendaciones son útiles en pacientes con dislipemias genéticas como LALD?
La LALD se ha tratado convencionalmente con estatinas para la dislipemia, que son eficaces para reducir la colesterolemia pero no evitan la progresión de la enfermedad hepática, y con trasplante ortotópico hepático para la hepatopatía evolucionada, en general con buenos resultados a corto y medio plazo. También se ha tratado con trasplante de células hematopoyéticas, pero hay poca experiencia y los resultados no han sido muy buenos.
Actualmente se dispone de la enzima LAL recombinante (sebelipasa alfa), un tratamiento enzimático sustitutivo muy eficaz y seguro, que se administra en infusiones intravenosas quincenales, y ya ha sido aprobado por la EMA y la FDA.
9. Una vez confirmado el diagnóstico. ¿Dónde pueden acudir los pacientes de este tipo de enfermedades? ¿Existe algún tipo de asociación?
Éstas pueden ser recomendadas:
⇢ FEDER (www.enfermedades-rars.org<http://www.enfermedades-rars.org/>)
⇢ AELALD (www.aelald.org<http://www.aelald.org/> / info@aelald.org<mailto:info@aelald.org>)


