Redacción Farmacosalud.com
Bajo el patrocinio de BIOGEN y NOVARTIS, www.farmacosalud.com acogió el webinar formativo ‘5 preguntas prácticas para el buen diagnóstico y abordaje de la AME (3ª edición)’. La sesión, dedicada por entero a la Atrofia Muscular Espinal (AME), contó con la moderación del Dr. Marcos Madruga, facultativo del Hospital Viamed Santa Ángela de la Cruz (Sevilla), mientras que como ponentes intervinieron los Drs. Jesús Eirís, neuropediatra del Hospital Clínico de Santiago de Compostela (La Coruña), y las Dras. Gema García Ron, pediatra del Centro de Salud La Rivota (Alcorcón, en Madrid), y María Calderón, neuropediatra del Hospital Virgen del Rocío (Sevilla). Eirís y García Ron respondieron a cuatro preguntas (dos preguntas para cada uno de los ponentes), mientras que la quinta fue contestada por Calderón. En su turno, el Dr. Eirís resaltó la relevancia de “pensar en CÓDIGO AME” así que se tenga la más mínima sospecha de estar ante un caso de tipo 1 o 2 de esta afección, puesto que sólo un diagnóstico temprano puede dar lugar a una intervención terapéutica temprana.
DR. JESÚS EIRÍS
1. ¿Cómo se diferencia el AME de otras enfermedades en edades tempranas?
Difusión: www.farmacosalud.com
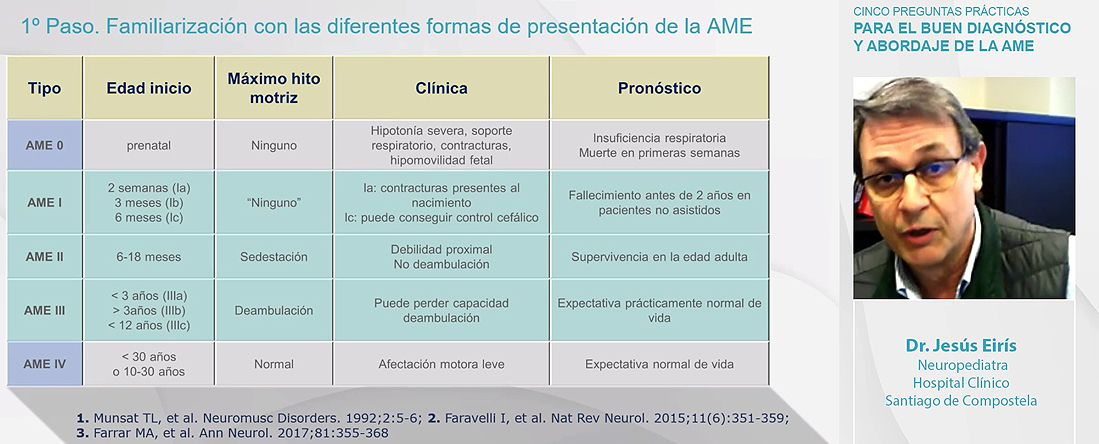
El AME tipo 1 se caracteriza por los siguientes signos:
-hipotonía y debilidad muscular de predominio en extremidades inferiores
-deficiente o ausente control cefálico
-disminución o ausencia de reflejos de estiramiento
-no sedestación y salvo para el tipo Ic tampoco un mínimo control cefálico
-posición de ‘rana’ o ‘libro abierto’
-deslizamiento en suspensión axilar (cuando se procede a la suspensión del niño a nivel de las axilas se observa deslizamiento porque no hay fuerza en la escultura escapular)
-actitud gravitatoria en suspensión ventral
-tórax en campana
-respiración paradójica (por producirse bamboleo abdominal)
-debilidad lingual y de musculatura deglutoria
-fasciculaciones linguales
-hipomimia con aspecto de alerta
AME tipo 2
-inicio de síntomas a los 6-18 meses de edad
-debilidad muscular progresiva con afectación más acusada en las extremidades inferiores, con hipotonía y arreflexia
-alcance de la sedestación pero no bipedestación
-con el crecimiento enfermedad pulmonar
-desarrollo de escoliosis
-contracturas articulares, anquilosis de la mandíbula, temblor y polimioclonus en las manos
-muy buena expresividad sin afectación cognitiva
Aspectos clave generales: a) no hay evidencia de alteración en el estado de alerta e interacción (muchos procesos neuromusculares cursan con afectación cognitiva o presentan trastornos del neurodesarrollo de distinta expresividad, cosa que no ocurre con el AME) b) hipotonía de características infrasegmentarias: hipotonía + hipoarreflexia + debilidad muscular c) curso progresivo de los síntomas. Salvo en tipo 0, no existe evidencia de enfermedad al nacimiento d) ausencia de rasgos dismórficos o malformaciones a otros niveles e) para diferenciar o acotar la etiología de AME, como datos de apoyo en laboratorio: CK normal o ligeramente elevada. Atención a los aumentos leves de CK ‘de transición’ (se eleva en las 2-3 primeras semanas de vida y después va normalizándose) f) como datos de apoyo electrofisiológico: patrón neurogénico en EMG (electromiografía).
“Lo más importante, lo que debemos tener en mente y lo que tiene que trascender, es que cuando formulemos una sospecha -aunque solamente se nos haya pasado por la cabeza- de que puede ser un AME", hay que proceder de inmediato a "pensar en CÓDIGO AME”, destacó el Dr. Jesús Eirís. Y es que únicamente un diagnóstico temprano puede dar lugar a una intervención terapéutica temprana.
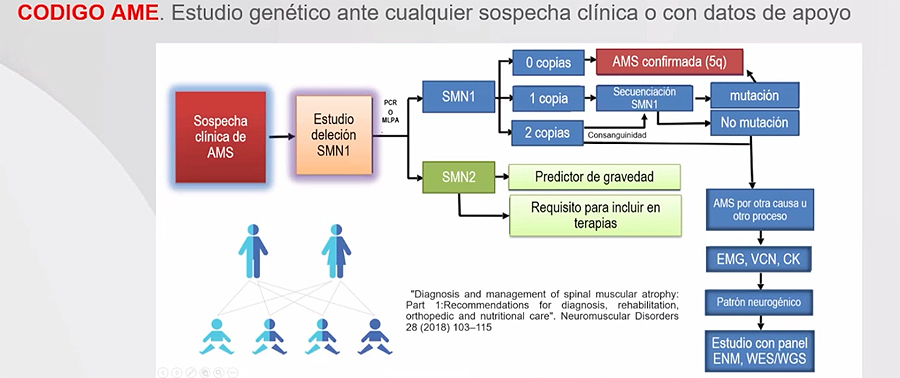
Se pueden obtener los resultados en 2 días
Difusión: www.farmacosalud.com
Difusión: www.farmacosalud.com
2. Y en edades tardías en el AME, ¿cómo se diferencia de otras enfermedades?
AME tipo 3
Los signos son similares a los anteriormente descritos, si bien se presentan de una manera más atenuada:
-debilidad muscular progresiva con predominio en extremidades inferiores
-alcance de la deambulación, pero pueden perderla evolutivamente
-hasta el 50% de las formas de inicio más tardío pueden permanecer ambulantes por encima de los 45 años. Característicamente, van a presentar marcha miopática (marcha de ánade), con basculación pélvica, hiperlordosis y signo de Gowers
-afectación de la musculatura respiratoria leve o inexistente
-no suelen desarrollar escoliosis grave
-puede existir temblor o polimioclonus en las manos
-la posible hipertrofia (pseudohipertrofia) de las pantorrillas puede llevar a confusión con otras patologías neuromusculares, en especial con la distrofia muscular tipo Becker.
El Dr. Eirís hizo hincapié en “el carácter progresivo” del tipo 3 de esta enfermedad, puesto que estos pacientes, hasta el momento en que se inicia la sintomatología de AME, han sido “niños totalmente sanos”. Tanto es así, que hasta entonces, hasta ese momento en el que empiezan sus síntomas, no han levantado “ninguna sospecha”, es decir, nadie ha imaginado que estén afectos de una patología neuromuscular que llegará a manifestarse con el paso del tiempo.
Para conocer el diagnóstico diferencial con otros procesos neuromusculares, acceder al vídeo que recoge la exposición del Dr. Eirís. Se puede visualizar la grabación clicando aquí o sobre la imagen que aparece bajo estas líneas:

Fuente: www.farmacosalud.com
Este webinar incluye vídeos con una clara finalidad médica y científica. Las imágenes que muestran niños con la patología tienen el consentimiento de difusión y emisión para este menester por parte de sus respectivos progenitores y están amparadas por la Ley de Protección de Datos


